Utilizing Fluorescence Microscopy To Characterize The Subcellular Distribution Of The Novel Protein Acheron Assignment Sample
SOLUTION
CHAPTER I - INTRODUCTION
Programmed Cell Death:
The genetic machinery needed to carry out programmed cell death, or cell suicide, is present in every cell (PCD). A variety of crucial functions are served throughout development and homeostasis by the successful beginning of PCD. The aetiology of human diseases including cancer, autoimmune disorders, and neurodegeneration, however, is driven by PCD dysregulation.
In 1842, Karl Vogt made the first-ever observation of cell death. Vogt identified cell death in the notochord and surrounding cartilage in metamorphic toads in his experiments (Vogt, 1842). Beginning with Dreisch's 1895 study of urchin embryo development and Spemann and Mangold's description of "organisers" in the African clawed toad Xenopus laevis, this ushered in a golden era of scientific discovery.
Vogt's discoveries weren't put in the "spotlight" however until almost a century later. In his article "Cell Death in Normal Vertebrate Ontogeny" published in 1951, Alfred Glucksman brought cell death back into the spotlight of developmental study. Glucksman describes his findings of the loss of the notochord core throughout development in this review. Richard Lockshin and Carroll Williams were two among the researchers that were motivated by these discoveries to continue their PCD study.
In the Journal of Insect Physiology between 1964 and 1965, Lockshin and Williams published a collection of five essays titled "Programmed Cell Death." The American silk moth Antheraea polyphemus undergoes eclosion at the conclusion of metamorphosis, and these studies investigated and addressed the neural, cellular, and hormonal processes influencing intersegmental muscle (ISM) cell death. These papers of biology assignment not only represent the first instances of the now-accepted phrase "Programmed Cell Death," but they also sparked a fresh surge of investigation into a hitherto understudied area. Sydney Brenner, Robert H. Horvitz, and John E. Sulston shared the 2002 Nobel Prize in Physiology or Medicine for their studies on the genetic control of organogenesis and programmed cell death in the Caenorhabditis elegans worm. These findings provide light on the crucial functions PCD performs in development and homeostasis. Over 70% of human illness is thought to be caused by deficiencies in PCD control (Reed, 2002). Numerous neurological conditions, including Parkinson's disease, in which striatal dopaminergic neuronal groups die, are caused by inappropriate activation of PCD (Hartman et al., 2000). Conversely, the root cause of many autoimmune disorders and the majority of malignancies is the inability to activate PCD to eliminate dangerous mitotically component cells (Renehan et al., 2001).
Apoptosis is the most well-understood method of cell death. Usually, a family of cysteine proteases known as caspases mediates this process (Miura et al., 1993). Numerous internal and external factors, including as pH changes, hormone exposure or loss, DNA damage, and loss of interaction with other cells, may cause apoptosis to occur (Norburry et al., 2001). Simply put, exposure to these death-inducing stimuli triggers a cascade effect in which effector caspase zymogens are activated by initiator caspases by interchain breakage. The executioner pro-caspases are then activated and cleaved as a result. Executor caspases then break essential proteins in the cell, causing apoptosis and cell death (Elmore, 2007). A closer look shows two primary mechanisms for initiating apoptosis in response to factors that cause death.
The first is the extrinsic route, which is started when members of the tumour necrosis factor (TNF) family of death receptors bind pro-death signalling peptides at the cell membrane (Micheau et al., 2003). Examples of this include ligands that bind to the Fas, DR4/5, and TNFR1 receptors, respectively, such as Fas-ligand (FasL), TNF related apoptosis inducing ligand (TRAIL), and Tumor Necrosis Factor Alpha (TNF) (Arai et al., 2014; Wiley et al., 1995; Cabal-Hierro et al., 2012). The recruitment of death domain/adaptors like Fas-associated protein with death domain (FADD) or Tumor necrosis factor receptor type 1-associated death domain protein is caused by the interaction of pro-death signalling ligands with TNF family pro-death receptors (TRADD). The death-inducing signalling complex (DISC) is created when these molecules are complexed (Scaffidi et al., 1999). Procaspase-8 may be recruited and activated by DISCs via death effector domains (Irmler et al., 1997).
Procaspase-8 is an initiator caspase that has the ability to cleave and activate executioner pro-caspases like caspase-3, -6, and -7, hence starting the PCD execution phase (Elmore, 2007). After then, apoptosis-related morphological and biochemical changes are brought on by activated executioner caspases (Slee et al., 2001). This includes the cleavage of crucial substrates including PARP (poly (ADP-ribose) polymerase) (caspase-3/7) and lamin A/C (caspase-6) as well as DNA fragmentation, nuclear collapse, and cleavage of chromatin (Wajant et al., 2002; Green and Llambi, 2015).
Initiating apoptosis via the intrinsic (mitochondrial) route is the second important process. Many of the same external and internal cues mentioned above may start this route, which is the main mechanism that initiates apoptosis in vertebrates (Bratton et al., 2001, Norburry et al., 2001). Members of the Bcl-2 (B-Cell Lymphoma 2) family of mitochondrial membrane proteins, as well as mitochondrial stress (oxidative stress)/mitochondrial outer membrane permeabilization (MOMP), have a significant impact on PCD in this pathway (Tait and Green, 2010). Three subfamilies may be created from bcl-2 proteins. Pro-apoptotic effector proteins from the first subclass include Bcl-2 associated X protein (Bax) and Bcl-2 homologous antagonist killer (Bak). For MOMP to occur, these proteins are both essential and sufficient (Moldoveanu et al., 2020). Anti-apoptotic regulatory proteins like Bcl-2, B-cell lymphoma-extra-large (Bcl-xL), or Mcl1 are part of the second subfamily (Tsujimoto, 1998). These proteins stop MOMP, which thereby stops cell death. The BH3-only proteins make up the third and last subfamily. These proteins include, but are not limited to, p53 upregulated modulator of apoptosis (PUMA), Bcl-2 interacting protein 3, Bcl-2-like protein 11, Bcl-2 associated agonist of cell death (Bad), and BH3 interacting-domain death agonist (Bid) (Hutt, 2015). By either inactivating antiapoptotic Bcl-2 proteins or activating proapoptotic Bcl-2 proteins, BH3-only proteins promote intrinsic pathway (Delbridge et al., 2016).
MOMP is produced directly by proapoptotic Bcl-2 proteins in the intrinsic cell death pathway (Eskes et al., 2000). Proteins like Bak or Bax oligomerize in the mitochondrial outer membrane after activation, creating holes in the outer membrane (Peng et al., 2009). This triggers MOMP, which releases proteins found in the mitochondrial intermembrane space such as cytochrome c, Smac/Diablo, and Omi (Goldstein et al., 2000; Munoz- Pinedo et al., 2006). Cytochrome C interacts to Apoptotic Protease-Activating Factor 1 (APAF1) after it has entered the cytoplasm (Zou et al., 1997). According to Kim et al. (2005), cytochrome c binding to APAF1 causes the cofactor's dATP to be hydrolyzed to dADP, which then enables the oligomerization of many APAF-1-dATP-cytochrome-c subunits to create an active apoptosome (Yu et al., 2005). Similar to the extrinsic cell death route, the intrinsic cell death mechanism uses caspase enzymes after the production of apoptosomes.
At the core of the apoptosome, Caspase-9 attaches to exposed caspase recognition domains (CARDS) (Yuan et al., 2010). A constant cycle of caspase-9 recognition, binding, activation, processing, and displacement is caused by the proximity of caspase-9 monomers (Malladi et al., 2009). Smac and Omi are released beforehand during MOMP and bind to and inhibit X-linked inhibitors of apoptosis (XIAP), which normally limit caspase-9 catalytic activity (Tait and Green, 2010). By using the ubiquitin proteasome pathway (UPP), XIAP causes caspases to be directly ubiquitinated and degraded (Eckelman et al., 2006).
Initiator caspase-9 and executioner caspases-3 and -7 become more readily available when XIAP is deactivated (Green and Llambi, 2015). Initiator caspase-9 then cleaves and activates executioner caspase-3 and -7, much as in the extrinsic route (Elmore, 2007). Executioner caspases then trigger cytoplasmic endonuclease and cytoskeletal peptide proteases, which may subsequently break down crucial nuclear proteins like lamin A/C and PARP (poly (ADP-ribose) polymerase), which are both catalysed by caspases 3/3/9. (caspase-6). (Green and Llambi, 2015; Wajant et al., 2002; Slee et al., 2001).
PCD of Manduca ISM:
Lockshin and William studied the loss of intersegmental muscles (ISMs) near the conclusion of metamorphosis in studies of the American silkmoth, Antheraea polyphemus. This mechanism was first identified in 1935 by Kuwana et al. and developed in 1956 by Finlayson. In order to exit their pupal cuticle at the conclusion of their pupal/adult development, adult moths use these ISMs. The next 30 hours saw the autophagic demise of these muscles (Finlayson, 1956; Beaulaton & Lockshin, 1977; Schwartz et al., 1993). The reasons behind this behaviour are still unknown, despite the fact that the phenomena of ISM loss in moths is widely understood.
The Schwartz team at UMass Amherst investigated the molecular processes behind this behaviour using the tobacco hawkmoth, Manduca sexta, as a model organism. ISM atrophy starts on day 15 of pupal development, according to research done in 1983 by Schwartz and Truman (Schwartz and Truman, 1983). The ISMs now go through hormonally-mediated atrophy, which is governed by a group of steroid hormones called ecdysteroids. During this period, they lose up to 20% of their volume and 40% of their muscular mass (Schwartz and Truman, 1983). The hormone responsible for insect moulting, 20-hydroxyecdysone (20E), was identified as the ecdysteroid in question. The expression of polyubiquitin mRNA and proteolysis both significantly rise during this phase (Schwartz et al., 1990a; Tsuji et al., 2020).
On day 17 of pupal development, a further drop in circulation levels of 20E triggers the production of the peptide hormone eclosion hormone (EH) (Schwartz and Truman; 1982). EH serves as the immediate cause of death. Rapid muscle atrophy after the release of EH is a defining feature of PCD.
(4% per hour after eclosion) mass (Schwartz and Truman 1983). Exogenous 20E administration the day before eclosion suppresses ISM PCD (Schwartz and Truman, 1984).
By showing that inhibitors of both RNA and protein synthesis might stop ISM PCD in Antheraea polyphemus, Lockshin expanded on the PCD phenomenon in 1969. (Lockshin and Williams, 1969). This provided as support for the hypothesis that de novo gene expression is necessary for PCD in silkmoths. Later, in 1990a, Schwartz et al. confirmed this. The Schwartz group was the first to clone genes linked to death by using the Manduca ISM model. Apolipophorin, 20S and 26S proteasome subunits, polyubiquitin, and other well-known compounds were among them (Schwartz et al., 1990b; Sun et al., 1995; Jones et al., 1995; Sun et al., 1997). Others, however, were brand-new proteins whose function in PCD was not yet known. This contains substances like Acheron, death-associated LIM-only protein, and small cytoplasmic leucine rich repeat protein (SCLP) (Hu et al., 1999; Kuelzer et al., 1999; Valavanis et al., 2007).
The induced sequence was dubbed Acheron (Achn) after one of the rivers of death in Greek mythology. Valavanis et al. (2007) used a differential cloning approach to discover genes that were induced or repressed when the ISMs were committed to die. When Manduca ISMs commit to dying on day 18 of pupal development, Acheron is drastically induced, according to Northern blot analysis of the mRNA expression of Acheron in Manduca ISMs (Valavanis et al., 2007). Following quantification using qPCR, it was shown that Acheron is stimulated by around 1,000 times on day 18. (Sheel et al., 2020). ISM death and Acheron expression were both slowed by 20E injection on day 17 of pupal development (Valavanis et al., 2007).
Acheron and PCD:
Acheron (LARP6), an RNA-binding protein belonging to the Lupus Antigen (LA) family with a 55kDA, is phylogenetically conserved. La-related proteins (LARPs) are RNA-binding proteins with a functional, winged helix-turn-helix structured RNA binding LA motif (LAM) and a well-conserved RNA recognition motif (RRM) (Stefanovic et al., 2014; Martino et al., 2015; Bousquet-Antonelli et al., 2009). A functional nuclear export signal (NES), subcellular localization and mRNA substrate recognition domain, and nuclear localization signal (NLS) are also present in Acheron (Valavanis et al., 2007; Stavraka et al., 2015).
Recent research by Dermit et al. indicates the significance of Acheron in the location and translation of ribosomal protein-coding mRNAs (RP-mRNAs). In order to improve Acheron localization and enrichment in actin-rich cell protrusions, Acheron also interacts with serine-threonine kinase receptor-associated protein (STRAP) through its SUZ-C domain. Here, Acheron interacts with RP- mRNAs, enhancing RP production, upregulating ribosome biogenesis, and increasing the amount of protein synthesised by migrating cells as a whole. Epithelial to Mesenchymal Transition (EMT) causes an increase in Acheron expression and subsequent protein production in human breast cancer (Dermit et al., 2020).
Intersegmental muscles (ISMs) from the moth Mnaduca sexta were used to create Acheron in its first cloning (references). Acheron expression is shown to rise 1000-fold on day 18 of pupal-adult development, when the ISMs commit to dying, using transcriptomins and qPCR (Sheel et al., 2020; Tsuji et al., 2020). When the quantity of 20E in the blood falls below a certain level in insects on day 17, the eclosion hormone receptor and the Acheron gene are both expressed (Sheel et al., 2020). On day 18, a further drop in 20E causes the release of eclosion hormone, which works on the muscles to promote the synthesis of cGMP, the transformation of inactive protein kinase-G (PKG) into active PKG (PKGa), and the phosphorylation of Acheron.
In parallel research, transcriptomics of developing Drosophila melanogaster showed that Acheron mRNA is momentarily increased by 600 times before adult eclosion (Graveley et al., 2010; Sheel et al., 2020). The results of the RNAi knockdown of Acheron expression in the muscles were muscle death before it should have, as opposed to after adult eclosion (Gurbutt et al., 2013, Sheel et al., 2020). Overall, the findings from Manduca and Drosophila point to a correlation between Acheron expression and muscle death during pupal development.
The interaction between Acheron and a pro-apoptotic BH3-only protein linked to BAD was discovered by mechanistic investigations of Acheron activity with Manduca ISM (Sheel et al., 2020). Acheron's ability to connect with a new 21 kDa BAD-like protein was revealed by Co-IP tests using day 18 ISM extracts of Acheron. When compared to Acheron, this protein's expression rose on day 18 of pupal development, according to a Western blot analysis, even though it was undetectable in the early stages of development. It's interesting to note that insects are said to lack BH3-only proteins (Nicolson et al., 2015). But computational RNA-seq study of the d18 Manduca ISM found a transcript transcribed at more than 10 RPKM that, when theoretically translated, was around 21 kDa and included patterns with similarity to BH3 domains (Sheel et al., 2020). This study led to the discovery of a new Bnip3 ortholog that shared multiple sequence regions with BAD (Sheel et al., 2020). The Schwartz Lab gave this molecule the moniker BBH-1 (BAD/Bnip-3 Homology 1). While RNAi of Acheron in the Drosophila ptelinal muscles led to the demise of valuable muscles and prevented the pharate adults from eclosing (Sheel et al., 2020).
In contrast, BBH-1 RNAi prevented muscle death, and the flies properly closed (Sheel et al., 2020). This shows that BBH-1 controls the Drosophila ecdysial muscles' ability to die directly.
Acheron interacts with the pro-apoptotic BH3-only protein, BAD, in mammalian models using Co-IP investigation of Acheron binding partners in C2C12 myoblasts, a presumed satellite cell line generated from adult mouse muscle and a common cell line used for studying myogenesis (Sheel et al., 2020).
Acheron binds to the ubiquitin ligase Human Homolog of Ariadne-1 (HHARI), according to co-IPs and yeast 2-hybrid screening (Wang et al., unpublished). The Parkin ubiquitin ligase, a recognised protein associated with autosomal recessive diseases, has no known near homolog, although HHARI does.
parkinsonism) (Aguilera et al., 2000). (Aguilera et al., 2000). Both proteins bind to the same ubiquitin conjugases (UbcH7 and UBcH8) and have comparable RING (Really Interesting New Gene) finger domains in common (Moynihan et al., 1999). Together, these findings imply that HHARI may serve as a ubiquitin ligase comparable to Parkin and be involved in controlling protein turnover (Marin and Ferrus, 2002).
Together, these results provide credence to the idea that Acheron's capacity to bind to poisonous BH3-only proteins (BAD in mammals and BBH-1 in insects) is a key component of its survival function (Sheel et al., 2020). The new pro-apoptotic protein BBH-1 is bound to by Acheron, which subsequently sequesters it, causing it to accumulate. Then, phosphorylated Acheron is likely destroyed by the Ubiquitin Proteasome Pathway (UPP), releasing BBH-1 in the process. Inhibiting or avoiding the activation of the anti-apoptotic Bcl-2 proteins, BBH-1 is subsequently thought to activate PCD via the intrinsic cell death pathway, resulting in MOMP (Sheel et al., 2020, Delbridge et al., 2016).
Acheron and Myogenesis:
The multifunctional protein Acheron takes involved in several developmental stages. The theory that Acheron is an essential regulatory molecule in the differentiation of both ciliated cells and muscles in mammals is supported by published research (Wang et al., 2009; Glenn et al., 2010; Manojlovic et al., 2017). In order to verify the idea that Acheron is essential for myogenesis, Wang et al. carried out immunofluorescence (IF), immunocytochemistry (ICC), Western blot, and caspase tests in both C2C12 myoblasts and zebrafish embryos in 2009. These studies' findings reinforced the idea that Acheron expression in C2C12 cells is developmentally controlled and that Acheron is a crucial component of myogenesis and may even control the survival of stem cells that are limited to the skeletal muscle. The aforementioned results also provide credence to the theories that Acheron controls the development of fast and slow twitch fibres in living cells and that Acheron causes reserve cells to die during differentiation.
The mouse cell line C2C12 is one of the model systems that has proven beneficial for analysing the expression and function of Acheron. These myoblasts are capable of committing to one of three probable destinies when they aren't given growth stimuli (Yaffe and Saxel, 1977). A minority of cells that follow the first route express the beta helix-loop-helix (bHLH) transcription factor MyoD (myoblast determination protein). This cell cycle arrest, myogenin expression, and differentiation into multinucleated myotubes are all results of this arrest (Thayer et al., 1989; Tapscott, 2005).
These cells' expression of the bHLH protein Myf5 opens up the second potential route (Myogenic factor 5). Myoblasts leave the cell cycle and arrest in G0 when Myf5 is expressed. Additionally, these cells increase their production of Bcl-2, an anti-apoptotic protein, making them more apoptosis-resistant (Dominov et al., 1998, 2001; Yoshida et al., 1998). Finally, a portion of cells fail to end the cell cycle or produce myotubes, and instead undergo apoptosis (Dee et al., 2002).
Acheron expression in C2C12 cells was shown by Wang et al. to be developmentally controlled (Wang et al., 2009). MyoD, a nuclear phosphoprotein that promotes the growth of multinucleated myotubes and drives myogenesis, was discovered by Western blot analysis of cycling and differentiated C2C12 cells (Tapscott et al., 1988; Wang et al., 2009). MyoD-expressing cells activate the lineage markers MyoA and MyoH when they lack growth factors. Proliferating myoblasts acquire differentiation signals from nearby cells after growth factors have been completely removed or exhausted. Acheron plays a crucial function in myogenesis by acting upstream of the muscle-specific transcription factor MyoD. Exogenous Acheron transfection caused cells to develop thicker muscle fibres with more nuclei and showed an almost total loss of satellite cells from the culture. Antisense Acheron transfected cells showed reduced myosin heavy chain (MHC), Myf5, and MyoD levels (Wang et al., 2009). An N-terminally shortened version of Acheron (tAchn), which seems to act as a dominant-negative regulator, was transfected into cells, and Western blot analysis showed that these cells produce MHC but not MyoD, high levels of Myf5, which indicates cellular arrest in G0 (Wang et al., 2009). Together, these results provide evidence for the hypothesis that Acheron is essential for myogenesis and cellular differentiation, and that blocking Acheron expression or activity prevents myogenesis.
Glenn et al. used immunofluorescence (IF) and migration tests using C2C12 myoblasts and myotubes to obtain insight into the molecular processes underlying Acheron's function in controlling myogenesis (Glenn et al., 2010). After being transfected with a plasmid encoding the full-length sequence of Acheron (FL-Achn), the transient activator of transcription (tAchn), or the antisense form of Acheron (AS-Achn), cells were examined for the presence of myogenesis-related proteins like MHC and both the A and B isoforms of the integrin subunit specific to laminin, 7A and 7B. Cells undergoing myogenesis need differentiation signals given by surrounding cells and the extracellular matrix in order to differentiate. Integrins are transmembrane proteins that control interactions (Boettiger et al., 1995).
After being transferred to differentiation medium (DM), cells expressing ectopic AS-Achn or tAchn prevented the production of 7A e, but cells expressing ectopic Acheron produce higher quantities of these Acheron proteins by the third day in DM (Glenn et al., 2010). As opposed to ASAchn- and tAchn-expressing cells, control and Achn-expressing samples were shown to have higher baseline levels of 7B protein and mRNA. In Achn-expressing and control samples after transfer to DM, 7B expression increased while remaining constant in ASAchn- and tAchn-expressing cells (Glenn et al., 2010). Myogenesis is increasing, while ITGB1, a crucial molecule in controlling cellular adhesion, is being downregulated, according to analysis of Acheron expression (Glenn et al., 2010). A migratory phase that peaks 24 hours after transfer to modified cells is indicated by increased regulation of 7A and 7B in the hours that follow (Glenn et al., 2010). An essential stage in cellular proliferation and adherence to extracellular matrix components is improved as a consequence, increasing cell to matrix contacts and adhesion differentiation (Beacham et al., 2006). (Beacham et al., 2006). When considered together, these data provide plausible methods for how Acheron-regulated myogenesis is mediated.
Hau et al. depleted Acheron mRNA from Xenopus embryos using morpholino oligos, much as the Schwartz lab's earlier zebrafish investigation. Following that, samples were examined using whole mount in situ hybridization and RT-PCR (WISH). These investigations' findings demonstrated that basal bodies at centrioles, which are necessary for ciliary assembly, were downregulated in the presence of Acheron knockdown, resulting in abnormalities in neural tube closure and a lack of cilia in the epidermis. Additionally, according to these scientists, Acheron regulates the expression of DNA synthesis-associated cell cycle protein (mcidas) and multicillate differentiation in a Notch-independent way. In Acheron knockdown cells, Mcidas stimulates the transcription of genes involved in centriole formation and may be utilised to partly restore multi-ciliated cell differentiation (Hau et al., 2020). These results provide credence to the idea that Acheron participates not only in myogenesis but also in the differentiation of ciliated cells.
Acheron and Pathogenesis:
A variety of illnesses, including cancer, muscle and neurodegenerative disorders, and AIDS, are linked to PCD dysregulation (Nagata et al., 2017; Reed et al., 1999). Since the three membrane growth factor receptors, ER, PR, and HER2/neu, which are the conventional targets for therapeutic intervention, are absent from triple negative basal-type breast cancer (TNBC), the prognosis for patients with TNBC is dismal (William et al., 2010). TNBC cell lines like MDA-MB-231 and Hs578T are often employed as cancer research models. Acheron expression was discovered to be higher in basal-type breast tumours compared to controls using microarray analysis and immunohistochemical tests on breast cancer tissues (Shao et al., 2012).
When implanted into immunodeficient SCID mice, MDA-MB-231 cells altered to produce ectopic Acheron developed tumours that were 5X bigger than non-engineered MDA-MB-231 controls (Shao et al., 2012). Additionally, ectopic Acheron-expressing tumours had higher levels of vascularization. This apparently caused proliferation to occur without being constrained by typical tumour development restrictions such food shortage and oxygen diffusion. Increased expression of the proteins matrix metalloproteinase 9 (MMP-9) and vascular endothelial growth factor were also seen in cells expressing Acheron, along with increased invasiveness through Matrigel (VEGF) (2012) Shao et al.
MMPs are created from inert zymogens. MMPs become active and break down the extracellular matrix (ECM) once the pro-domain or residue modification is removed by proteolysis (Kessenbrock et al., 2010). This behaviour is physiologically controlled by factors including immunological reactions and inflammation (Kessenbrock et al., 2010). After activation, MMPs degrade the ECM around tumours that are actively growing. Due to the resulting cell migration and subsequent inflammation, a positive feedback loop to release additional MMPs is set off. MMPs are one of the primary indicators of aggressive malignancies as a result. When compared to wild type cell lines, MDA-MB-231 cells with MMP-9 knockdown (KD) showed significantly reduced levels of Matrigel invasion. Tumors with decreased MMP9 expression developed when transplanted into mice, however unlike WT tumours, these tumours were unable to spread to the animals' lungs (Mehner et al., 2014).
By causing the release of activated vascular endothelial growth factor-A (VEGF-A) and vascular endothelial growth factor receptor 2 (VEGFR-2), MMP-9 differs from other MMPs in that it further improves angiogenic activity (Shibuya et al., 2011). By attaching to VEGFR-2, VEGF-A promotes the proliferation of endothelial cells. As a result, tumour vascularization, tumour growth, and cell proliferation are all improved. Three human cancer indicators that significantly enhance disease mortality (Wang et al., 2018).
TNBCs have a greater capacity for invasiveness and angiogenesis.
Human ductal carcinoma immunohistochemical labelling demonstrates that Acheron expression is increased in malignant tumours that are aggressive (Shao et al., 2012). TNBCs with ectopic Acheron expression have increased invasiveness and angiogenic capacity. Together, these facts provide credence to the idea that Acheron is essential for the pathogenesis of TNBC.
The pathophysiology of the liver may also be influenced by Acheron dysregulation. The illness fibrosis is characterised by increased amounts of type I collagen in different organs (Ghosh et al., 2002).
Acheron regulates the subcellular location and expression of type I collagen mRNAs by binding to their 5' stem loop (5'SL) (Cai et al., 2010). Loop 3 of Acheron's RNA Recognition Motif (RRM) binds to the 5' SL and sequesters the complex to proteins like the protein transport protein SEC61 translocon, causing this to happen (Stefanovic et al., 2014). Following the C-terminal phosphorylation of Acheron, type I collagen production takes place. At the C-terminus, there are six serine residues, all of which are phosphorylated in a hierarchical manner to promote the production of type I collagen. This starts with the phosphorylation of S451 by Protein Kinase B (Akt/PKB), and continues with the phosphorylation of S348 and S409 by the Mammalian Targets of Rapamycin Complex 1 (mTORC1). Type I collagen production is hindered or decreased as a consequence of improper control of phosphorylation at these locations or suppression of Akt and mTORC1, respectively (Zhang et al., 2016, 2017). Immunosuppressive medications like Tacrolimus (FK506) that disrupt the Acheron/collagen mRNA binding restrict the Acheron to 5'SL binding and significantly lower the production of type I collagen (Manojlovic et al., 2013).
A dynamic protein known as acheron, it is essential for PCD, myogenesis, malignancy, and hepatic fibrosis. Acheron has functions in additional illnesses that have not yet been fully investigated, despite the fact that these two disease pathways have been identified. A characteristic of type 2 diabetes' pathogenesis, greater proinsulin levels are also linked to acheron (Strawbridge et al., 2011).
By examining Acheron's capabilities to serve as an RNA binding and signal transduction protein, it becomes clearer how it may participate in a variety of crucial physiological activities. The multidomain scaffolding protein calcium/calmodulin-dependent serine protein kinase 3 (CASK) may influence ion channels, receptors, and cell adhesion molecules in epithelial tissues, motor neurons, and muscles to help regulate signalling (Dimitratos et al., 1998). CASK binds to the C-terminus of Acheron through its CaM K II-like domain, according to a yeast 2 hybrid screening experiment using Acheron as the bait and a mouse embryonic cDNA library as the prey (Weng et al., 2009). The complexation of CASK and Acheron with transcriptional repressors of the helix-loop-helix (HLH) family known as Id (inhibitor of DNA binding/differentiation) allows for the control of gene expression (Hsueh, 2006; Weng et al., 2009).
Through RNA and protein-protein interaction, Acheron can operate as a crucial regulatory molecule thanks to its functioning RRM, La Motif, and RBD. Acheron binds to the 5' stem-loop structure (SL) of type I collagen mRNA to control the production of type I collagen. Translation of the collagen mRNA is enabled by Acheron's binding to the 5'SL (Fritz et al., 2011). When structural proteins like Myosin IIB and Vimentin join forces to create the Acheron complex, it binds to the 5'SL of type I collagen and sequesters it to the ER membrane (Wang et al., 2014; Cai et al., 2010; Manojlovic et al., 2012). Then, complexed Acheron attaches to the C-termini of RNA Helicase A (RHA) and Serine-Threonine Kinase Receptor-Associated Protein (STRAP), enabling translation (Vukmirovic, 2013; Manojlovic et al., 2012).
A dynamic chemical called acheron has important regulatory roles in PCD, myogenesis, and pathogenesis. Acheron action is governed by protein-protein interactions and its subcellular localization patterns, according to a number of lines of evidence, despite the fact that the mechanisms governing its action are poorly understood. In this dissertation, I will use immunofluorescence and confocal microscopy to carry out two studies that may shed light on Acheron's functions in mammalian models of programmed cell death, differentiation, and pathogenesis in addition to revealing its subcellular distribution and potential binding partners.
It is generally known that comprehending a protein's subcellular localization may assist explain both the structure of the cell as a whole and the function of an individual molecule (Scott et al., 2005). A dynamic protein, Acheron plays mechanistic roles in processes within and outside the nucleus, such as differentiation, programmed cell death, and disease, respectively. It has functional nuclear localization and nuclear export signals (Valavanis et al., 2007, Sheel et al., 2020). Acheron's precise subcellular location remains uncertain, nevertheless. I initially investigated the idea that Acheron localises to mitochondria because it interacts with BAD, a protein directly connected to MOMP.
Protein-protein interactions may have a role in Acheron's participation in PCD, myogenesis, and pathogenesis, according to available evidence. I'll investigate the claim that the protein Acheron colocalizes with the molecules BAD, HHARI, and calcium/calmodulin-dependent Serine Protein Kinase (CASK).
CHAPTER II
DETERMINING THE LOCALIZATION OF ACHERON AND SPECIFIC BINDING PARTNERS IN A MAMMALIAN SYSTEM
Introduction:
The multifunctional protein Acheron possesses various protein-protein interaction motifs that are likely responsible for many of its biological functions. Even while new information on the subcellular processes that control Acheron function has emerged, there are still many unanswered questions.
.png)
.png)
Table 1a. Known Acheron Binding Partner Roles, Localization and Evidence:
Listed here is a list of recognised Acheron binding partners with established involvement in PCD, myogenesis, and pathogenesis. The location of each molecule and the information available describing its interaction with Acheron are also given in the table along with each molecule's physiological function.
I used an anatomical approach to test three main assumptions in order to understand some of the molecular processes underlying Acheron function in mammalian systems. The first investigation established Acheron's subcellular location in mammalian cells. Here, I assessed how well Acheron adhered to the various mitochondria, actin filaments, and microtubules. To identify Acheron's cellular location, cycling mononucleotide C2C12 murine myoblasts, differentiated C2C12 my tubes, and U2-OS osteosarcoma cells were immunostained. The second research examined the idea that Acheron's binding partners' homologs locate alongside Acheron in mammalian cells. In this investigation, HHARI, BAD, and CASK in relation to -tubulin were stained on Wild-type C2C12 myoblasts and U2-OS osteosarcoma cells. The third and final experiment explored the possibility that CASK moves to the nucleus at the same time that Acheron does in response to growth factor stimulation. To test this theory, C2C12 cells were cultivated on glass coverslips with ectopic full-length GFP-tagged Acheron and C2C12 cells with ectopic full-length GFP-tagged Acheron that had deletions in their nuclear export signal (- NES). High concentrations of growth stimulants were then used to push Acheron into the nucleus, where it was trapped in -NES cells. The position of Acheron and CASK in regard to microtubules was then labelled in cells, and any translocation was examined.
Materials and Methods
C2C12 Cell Culture:
In DMEM (Dulbecco's Modified Eagle Medium) + 10% FBS (Fetal Bovine Serum) + 1% penicillin/streptomycin in 10% CO2, C2C12 cells were cultured at 37°C. A new growth media was made by vacuum filtration in a tissue culture hood at 25°C using 445 mL DMEM pH 7.2, 50 mL FBS, and 5 mL Pen/Strep. The cells were then taken out of liquid nitrogen storage and promptly defrosted by rotating the tube in a water bath that was maintained at 37°C. Vials were transferred to 15 mL conical tubes with 5 mL of full C2C12 DMEM media after being 70% thawed, sterilised with ethanol, and then centrifuged at 500 x G for 5 minutes.
After discarding the supernatant, the pellets were reconstituted in 10 mL of full C2C12 DMEM media and then plated on 10 cm plates with flat bottoms. After that, cells were divided every 48 hours (or when they achieved around 70% confluency) by aspirating the medium and twice rinsing them in PBS (phosphate buffered saline). The cells were then added to 0.53mM EDTA + 0.25% (w/v) trypsin that had been warmed to 37°C. Cells were once again transferred to a 15mL conical after being removed from the dish's bottom and centrifuged at 500 x G for 5 minutes. The pellets were then resuspended in 10 mL of full C2C12 DMEM media after the supernatant was discarded. Then, cells were plated in a 10 cm flat-bottom dish at a ratio of 1:10 of cells to new media.
C2C12 Differentiation:
C2C12 cells were cultivated to a confluency of around 70% before being moved to a low serum differentiation medium (DM) composed of DMEM + 1% Pen/Strep + 2% horse serum. The differentiation medium was changed every 48 hours while the cells were cultured in it for 5 days at 10% CO2 37°C. For effective differentiation, the development of myotubes in the cells was seen under a microscope.
Glass Coverslip Coating Removal and Poly-L-Lysine Treatment:
Glass coverslips (Fisher Scientific) were treated for 15 minutes with 21 mM Poly-L-Lysine (Sigma Aldrich) to improve cell-to-coverslip adhesion. Coverslips were taken out of their packing, put on the shaker, and spun at 100 rpm for six hours at 25 °C in 25 mL of 2M NaOH.
After that, coverslips were submerged in 20 mL of dH2O and shaken for a whole night at 25 °C at 150 rpm. The next day, coverslips were washed in running dH2O before being sterilised in the autoclave. After being washed with 70% ethanol, sterile coverslips were positioned in the tissue culture hood. Coverslips were coated with 10 mL of 21 mM Poly-L- Lysine and left in the tissue culture hood for 15 min at 25 °C. Coverslips were then rinsed with Poly-L-Lysine solution, aspirated, and overnight air dried in the tissue culture hood with the filtration on. The resulting coverslips were deemed sterile and suitable for cell culture.
C2C12 + HA-Acheron -NES / +NES Mutant Acheron Translocation:
For the studies, ectopic Acheron with a HA-tag was expressed in C2C12 cell lines. Cycling C2C12 cells were transfected with a plasmid that used a CMV promoter to drive ectopic Acheron expression. The Nuclear Localization Signal, NLS (-NLS), or the Nuclear Export Signal, NES (-NES), were then deleted from constructs using site-directed mutagenesis and injected into cells (Shao et al., 2012). We used mutant cells with malfunctioning NESs and cultivated them at high growth serum to "push" Acheron towards the nucleus in order to ascertain Acheron's function there (Glenn and Schwartz, unpublished). The cells were grown in conventional C2C12 growth media (10% FBS in DMEM) for 24 hours on glass coverslips at 37 °C and 10% CO2. The cells were then transferred back to high growth serum medium (15% FBS) for 2 hours after spending the previous night in 3 mL DMEM + 1% Pen/Strep + 2% horse serum. The medium was aspirated once the cells were taken out of the incubator. Following a 1X PBS washing, the cells were fixed by adding 2 mL of 4% paraformaldehyde to 1X PBS at 4°C and incubated for 10 to 15 minutes at 25°C.
Paraformaldehyde was fixed, then removed and disposed of as hazardous waste. Dishes were sealed with parafilm and coated with 1X PBS to keep them at 4°C until they were ready for immunofluorescence staining.
Nocodazole Mediated Microtubule Depolymerization:
Nocodazole (Sigma Aldrich), an anti-cancer drug, was used to treat C2C12 cells in order to cause microtubule depolymerization and sabotage connections with microtubule-associated proteins (MAP) (Verstraelen et al., 2017). C2C12 cells were grown on glass coverslips coated with Poly-L-lysine in flat bottomed 6-well plates in GM at 37 °C with 10% CO2. The media was aspirated and new growth medium was added after the cells reached 70% confluence. Then, nocodazole was immediately added to each well, and it was incubated for an hour at 37°C with a final concentration of 500nM. Figures 6a and b show the empirical results of the concentration determination.
C2C12 and U2OS: Acheron, BAD, Ariadne, CASK, E7 Tubulin Colocalization
Immunofluorescence Staining:
C2C12 cells were stained after being cultivated in GM at 37 °C and 10% CO2 on glass coverslips coated with poly-L-lysine. Dishes were taken out of the incubator once the cells reached the required confluency and surface sterilised with 70% ethanol. After that, dishes were brought into the tissue culture hood. Each well's remaining medium was aspirated, and the cells were then washed with 1X PBS pH 7.2 until the medium's red/pink colour was gone. Then, after being incubated at 25°C for 10-15 minutes, the cells were fixed by adding 2 mL of 4% paraformaldehyde pH 7.2 in 1X PBS at 4°C. Dishes were sealed with parafilm and coated with 1X PBS to keep them at 4°C until they were ready for immunofluorescence staining.
Samples were taken out of the refrigerator and given a fresh 1X PBS pH 7.2 wash at 25°C for 5 minutes before to the immunofluorescence staining. Then, slides were put via a to improve fluorescent signal and reduce autofluorescence, methanol permeabilization step is used. 100% methanol was stored overnight at -20°C. After being cleaned, slides were put on ice and coated with 100% ice-cold methanol. To avoid dehydration, coverslips were immediately put to -20°C for 8 minutes. Existing methanol was eliminated from each slide after 8 minutes. After that, slides were washed on the shaker at 300 rpm for 5 minutes while being submerged in brand-new 1X PBS at 25°C.
Coverslips containing cells were first permeabilized with methanol, washed, and then incubated in an immunofluorescence blocking solution. During the methanol permeabilization process, blocking solution was freshly prepared by combining 5% goat serum (ThermoFisher Scientific) in 1X PBS at 25°C and mixing by inversion. Following that, coverslips were incubated in 1 mL of immunofluorescence blocking solution overnight at 4 °C with 200 rpm of the shaker. Parafilm was also used to wrap dishes at this period to stop slides from drying out.
The next day, each coverslip received a 1mL fresh primary antibody dilution solution in lieu of the blocking solution. We prepared the primary antibody dilution solution by mixing 2% BSA (Sigma Aldrich) diluted in 1X PBS at 25°C with 1X PBS pH 7.2. The appropriate primary antibodies were then applied to the appropriate slides at the following dilutions, and the slides were shaken at 200 rpm at 4°C overnight to incubate them. To avoid coverslip dehydration, plates were once again covered with parafilm during this period.
1. E7 (DSHB) Anti-Beta Tubulin, 1:350 l
2. LARP6 (Schwartz Lab Purified) Anti-Acheron, 1:1000 mL.
3. Anti-Ariadne: 1:5000L of HHARI1 (Schwartz Lab Purified).
4. Anti-BAD and BAD (Abcepta) - 1:500 l
5. Anti-BAD and BAD (LSBio), 1:500 l
6. CASK (ABGENT) - 1:500L Anti-CASK C-Terminus
After incubation, dilution solution was withdrawn from each well, and slides were then washed three times in 1X PBS pH 7.2 at 25°C for 20 minutes while being shaken at 350 rpm. Fluorescent dyes were then applied to each individual slide at the following dilutions after the slides had been coated with 1mL of fresh 1X PBS pH 7.2. After that, slides were hidden from view and incubated in PBS/fluorescent dye solutions for 1.5 hours at 25°C.
1. Nuclear stain DAPI, 1:300 mL
2. Dylight 594-1:500L
3. Alexafluor 647, 300 L
4. Alexafluor 488 - 0.5 mL
5. Mitotracker CMXRos 40 m
6. 1:1000 L Rhodamine Phalloidin
Following the removal of the secondary antibody solutions from each slide, the slides were once again washed three times on the shaker at 350 rpm in 1X PBS pH 7.2 at 25 °C for 20 minutes. After that, coverslips were taken out of the wells and mounted using 6.5 l of immunofluorescence mounting media face down on labelled slides (4:1 glycerol in PBS). Before imaging, slides were allowed to dry overnight at 4°C with the edges sealed with clear nail polish to avoid coverslip dehydration. After that, slides were processed in Fiji and photographed using a Nikon A1R-SIMe resonant scanning confocal super resolution microscope.
Results
Acheron Localization in Mammalian Cells:
Cycling murine C2C12 myoblasts, differentiated C2C12 myotubes, and human U2-OS osteosarcoma cells were treated for immunofluorescence labelling of Acheron in order to ascertain the subcellular location of this protein in mammalian cells. Areas of colocalization between Acheron (green) and other markers (actin, mitochondria, and tubulin) (red) may be deduced from the resultant yellow signal throughout these tests.
Co-IP of Acheron in C2C12 myoblasts revealed binding to the BH3-only protein BAD, which is typically found localised in mitochondria (Sheel et al., 2020). (Jiang et al., 2006). Mononucleated C2C12 myoblasts were stained for Acheron (green), nuclei (DAPI (4′, 6-diamidino-2-phenylindole/DNA fluorescent dye) (blue), and mitochondria (Mitotracker CMXRos) (red), and imaged at 60X on a Nikon Eclipse TE2000 inverted fluorescent microscope to test the hypothesis that Acheron localises to mitochondria with BAD in some circumstances. The bulk of the Acheron does not colocalize with the mitochondria, despite the fact that tiny quantities of yellow signal may be observed in the cell's perinuclear regions (Figs 1a, b). Instead, the cytoskeleton and nucleus seem to be the primary sites of Acheron expression. The fluorescence intensity in arbitrary units (a.u) is shown versus the cell's diagonal length in Figure 1b (microns). Simply put, this graph shows how bright each pixel is in relation to other parts of the cell. The Acheron and Mitotracker intensities are shown as green and red lines, respectively, while DNA staining is shown as the blue line. The cell nucleus may be precisely identified by the increase in DAPI (blue) fluorescence. This picture thus shows us where the majority of the Acheron is located inside the cell (Figure 1a). Acheron (green) and Mitotracker (red) peak may be detected outside the nucleus in the graphic (blue). Our perinuclear colocalizing signal, most likely. Areas of DAPI fluorescence may be observed overlapping with a large portion of the remaining Acheron signal (blue).
This shows that a significant portion of the Acheron dwells in the nucleus of this cell, whereas the other components are either present perinuclearly or in the cytoplasm of the cell (Figure 1b). The staining seen in Figure 1c lends credence to this. Acheron staining with higher resolution reveals that the cytoplasmic, non-nuclear Acheron signal is not uniform but rather seems to follow a particular cytoskeletal structure (Figure 1c). We can discern an unique reticulated pattern in our perinuclear Acheron signal thanks to the improved clarity of the Acheron staining in this Figure. Along the border of the nucleus, Acheron concentrations may be detected. Acheron may also be observed moving in a specific pattern away from the inner membrane but still close to the nuclear envelope's edge and farther inside the cell. These are probably the rough endoplasmic reticulum and the perinuclear region, respectively. Small Acheron aggregates may also be detected at the cell's edge, however they are clearly on protrusions that stick out from the cell rather than the leading edge. This most likely affects filopodia rather than lamellipodium (Figure 1c). This gave us the impression that Acheron was concentrating in the cytoskeletal components of the cell.
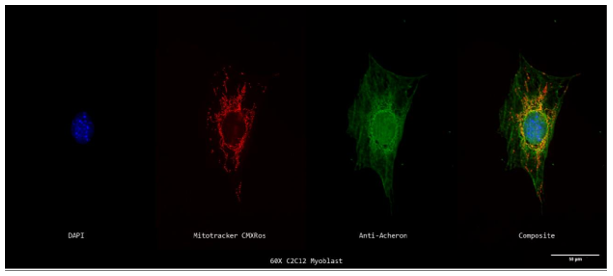
Figure 1a.: Subcellular Localization of Acheron relative to Mitochondria in C2C12 Myoblasts
A 60X z-projection of a single stained cycling mononucleotide C2C12 myoblast's nucleus (DAPI; blue; far left), mitochondria (Mitotracker; red; second to the left), Acheron (anti-Acheron; green; second to the right), and composite (merge; far right) channels. Some
While the majority may be seen in the cell's nucleus or cytoplasm, Acheron can be observed localizing with mitochondria in the yellow sections of the picture.
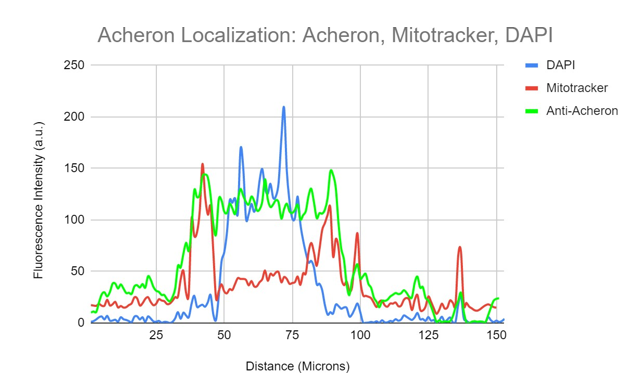
Figure 1b. Colocalization analysis of DAPI, Acheron and Mitochondria fluorescent signal from figure 1a.
An illustration of the relationship between fluorescence intensity for DAPI(blue), mitochondria (red), and anti-Acheron (green) and cell area. However, substantially more of the Acheron signal can be seen overlapping with the DAPI (blue) signal at the cell's nucleus and in its cytoplasm. Acheron (green) fluorescence intensity can be seen overlapping with mitotracker (red) in areas of perinuclear signal.
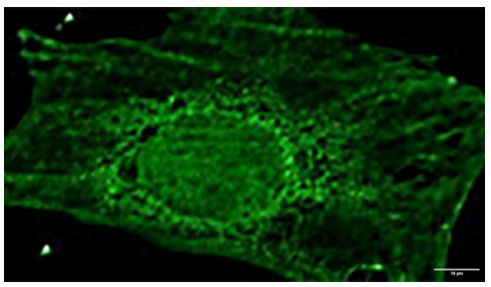
Figure 1c. Acheron Staining Follows a Cytoskeletal Pattern in C2C12 Myoblasts.Figure 1a's Acheron (green) staining as seen up close. Instead of being diffuse, the cytoplasmic signal seems to follow a cytoskeletal structure.
Cycling C2C12 myoblasts were labelled for Acheron (green), DNA (DAPI; blue), and actin (Rhodamine Phalloidin; red) and photographed at 60X using a Nikon Eclipse TE2000 inverted fluorescence microscope to test the theory that Acheron binds to actin filaments (Figure 2a). In this image, there are no colocalization regions (yellow), which suggests that Acheron does not bind to actin filaments. However, Acheron is once again detected accumulating towards the actin filament edges (likely the filopodia). Additionally, the acheron signal is more diffuse in the nucleus and the region around it, but pattern-like in the cytoplasm, suggesting that there may be various molecular actions depending on protein location (Figure 2a). The information from Figure 2a's graphs enables one to reach the same conclusions. Rhodamine phalloidin (red) fluorescence and Acheron (green) signal both exhibit cytoskeletal expression patterns that are comparable, but the signals do not coincide, indicating that Acheron (green) does not localise to Actin (red) filaments in the cell body. However, similar to Figure 1b, Figure 2b also exhibits a spike in Acheron (green) expression at the periphery. Probably, Acheron's enriched region is located here.
Confined to the cell's filopodia. Even while this confirms the findings discovered by Dermit et al., the idea that Acheron attaches to actin filaments is not supported by the lack of yellow fluorescence (Figure 2b). As a result, we focused on microtubules, another cytoskeletal component.
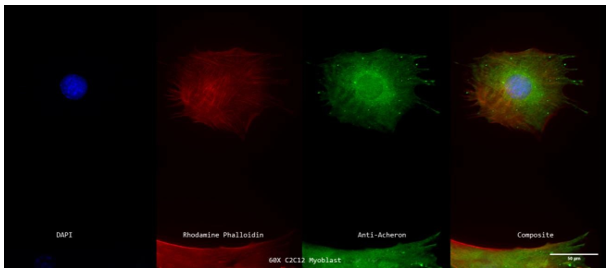
Figure 2a: Subcellular Localization of Acheron Relative to Actin Filaments in C2C12 Myoblasts.
A 60X z-projection showing a single stained cycling mononucleated C2C12 myoblast's nucleus (DAPI; blue; far left), actin filaments (Rhodamine Phalloidin; red; second to the left), Acheron (anti-Acheron; green; second to the right), and composite (merge; far right) channels. Acheron may not bind to actin filaments as there are no regions of apparent colocalization (denoted in yellow). Acheron may still be recognised, nevertheless, in the cytoplasm and nucleus in a cytoskeletal pattern.
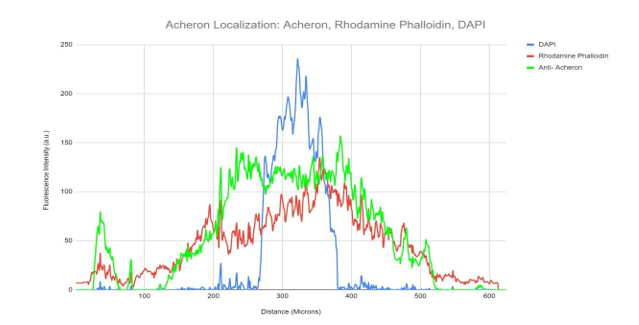
Figure 2b. Colocalization analysis of DAPI, Acheron and Rhodamine Phalloidin Fluorescent Signal From Figure 2a.
A graphical representation of fluorescence intensity for DAPI (blue), Rhodamine Phalloidin (red) and Anti-Acheron (green) plotted relative to area on the cell. Acheron (green) fluorescence can be seen following a separate staining pattern from Rhodamine Phalloidin signal (red).
We stained cycling C2C12 myoblasts for Acheron (green), DNA (blue), and -tubulin (red) to examine the possibility that Acheron attaches to microtubules (Figure 3a). Microtubule formations are made up of the tubulin monomers -tubulin and -tubulin, which polymerize together (Gunning et al., 2015). A significant amount of overlap was found between the Acheron and -tubulin signals (yellow), indicating that the bulk of the Acheron protein (green) may be seen colocalizing with -tubulin (red) on microtubules (Figure 3a). Similar to earlier Acheron labelling, green, fluorescent signal is seen at cell protrusions, which are probably actin-rich filopodia. In Figure 3b, the precise level of colocalization is visually shown. This graph also shows fluorescence intensity (a.u.) graphed versus the diagonal length of the cell, similar to Figure 1b (microns). The blue line in this diagram represents DNA, whereas the green and red lines, respectively, in this diagram indicate Acheron and -tubulin. even when the DNA signal is the Acheron and -tubulin signals localised to the nucleus of the cell are detected fluorescing at different intensities but at roughly the same places throughout the cell.
One region outside the nucleus also experiences an increase in acheron and -tubulin signal (Figure 3b). This is most likely the centrosome, a cellular component essential to microtubule organisation (Bornens and Azimzadeh, 2007). The aberrant spikes of Acheron (green) signal at the cell's edge serve as a visual representation of the Acheron signal aggregates found in cell protrusions. Together, these findings lend credence to the idea that Acheron localises to microtubules.
Rather than using a Nikon Eclipse TE2000 inverted fluorescent microscope, cycling C2C12 myoblasts were labelled for Acheron (green), DNA (blue), and -tubulin (red) and photographed at 100X using a Nikon A1R- SIMe resonant scanning confocal super resolution microscope (Figure 3c). Confocal microscopy employs a spatial pinhole to exclude out-of-focus light during image capture, which sets it apart from traditional inverted fluorescence microscopy. The optical resolution and contrast of microscopic samples are greatly improved. Scanning confocal microscopes may also automatically take a number of two-dimensional pictures within a predetermined focus range. The "z-slices" that are produced may be used to recreate a three-dimensional structure. When two proteins are believed to be simultaneously occupying the same location, these methods become crucial for co-localization research. Precision co-localization analysis is made possible by the ability to conduct protein localization analysis in z-slices up to 0.125 microns thick (Pawley, 2006; Jensen, 2004). Five 0.125-micron thick z-slices, or a roughly 0.625-micron thick z-stack, were taken from the three-dimensional reconstruction of this picture and processed separately. All of the microtubule and acheron in-focus slices are present in this region of the cell, free from interference from the optical slices above or below. Because of the removal of optical "contamination," resolution is improved (Figure 3c). Again, extensive areas of yellow fluorescence throughout the cell indicate colocalization of both proteins.
Returning to the centrosome to assemble. Additionally, similar to earlier photos, strong clusters of green (Acheron) fluorescence can be detected along the cell's edge. This provides further evidence in favour of our claim that Acheron binds to microtubules. At the edge of the cell, patches of distinctly red microtubules can also be observed in this view. We are not given this level of detail by conventional inverted fluorescence microscopy.
When the same information is again examined visually, it becomes even more clear. Pictures obtained using confocal super resolution microscopy include thousands of data points, enhancing the temporal resolution compared to images obtained using ordinary inverted fluorescence microscopy, which only have hundreds of data points. Here, the Acheron (green) and -tubulin (red) signals perfectly match each other. Additionally, the Acheron signal is unaffected by peaks in the DAPI (blue) fluorescence (Figure 3d).
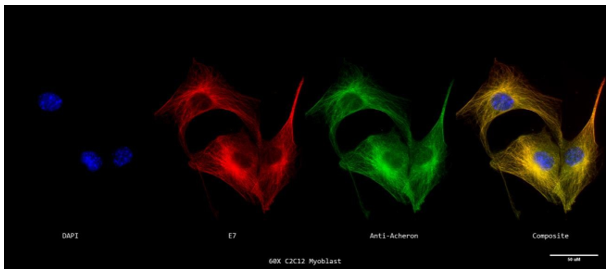
Figure 3a. Subcellular Localization of Acheron Relative to Microtubules in C2C12 Myoblasts.
A z-projection of the stained cycling mononucleated C2C12 myoblasts' nuclei (DAPI; blue; far left), microtubules (E7; red; second from the left), Acheron (anti-Acheron; green; second from the right), and composite (merge; far right) channels is shown here. Smaller concentrations of Acheron are shown localising to the cell's nucleus, while larger concentrations are visible colocalizing with microtubules in the yellow portions of the picture and becoming more intense near the cell's centrosome. This suggests that Acheron and -tubulin have a high colocalization.
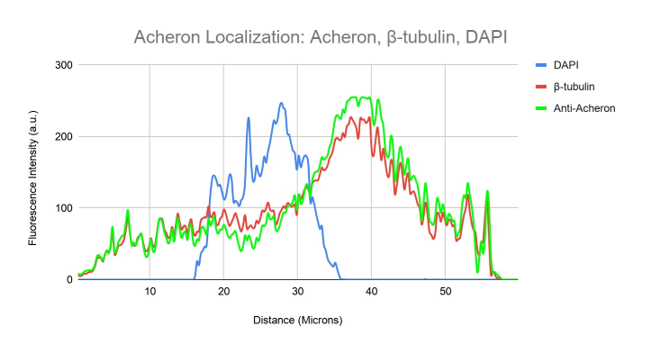
Figure 3b. Colocalization analysis of DAPI, Acheron and β-tubulin Fluorescent Signal From Figure 3a.
DAPI (blue), -tubulin (red), and Anti-Acheron (green) fluorescence intensity is shown on a graph in relation to cell surface area. Acheron's (green) and -tubulin's (red) fluorescence intensities nearly exactly overlap, showing that both proteins may be detected on the same regions of the cell while having differing levels of expression.
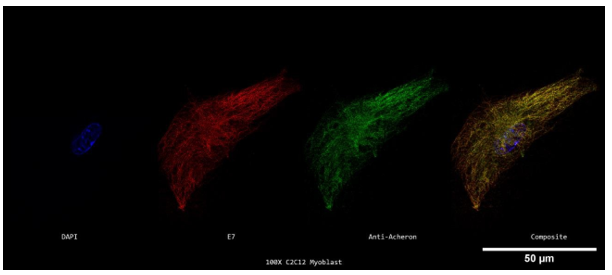
Figure 3c. Super Resolution Subcellular Localization of Acheron Relative to Microtubules in C2C12 Myoblasts.
A 100X confocal z-stack of a single labelled cycling mononucleated C2C12 myoblast's nucleus (DAPI; blue; far left), microtubules (E7; red; second to the left), Acheron (anti-Acheron; green; second to the right), and composite (merge; far right) channels. A little amount of Acheron can be detected in the cell's nucleus, but the majority of it can be observed colocalizing with microtubules in the yellow portions of the picture, with greater patches of yellow fluorescence in the cell's centrosome.
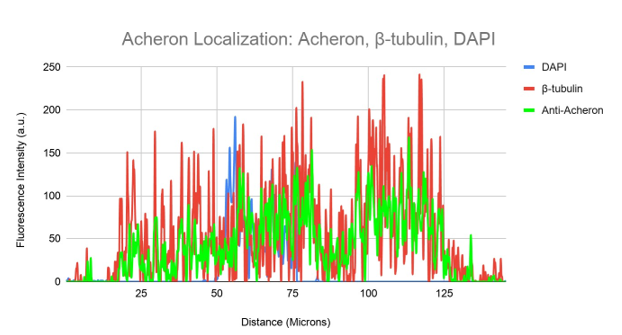
Figure 3d. Colocalization analysis of DAPI, Acheron and β-tubulin Fluorescent Signal From Figure 3c.
DAPI (blue), -tubulin (red), and Anti-Acheron (green) fluorescence intensity is shown on a graph in relation to cell surface area. While DAPI fluorescence is still largely distinct, the intensity of the anti-Acheron (green) and -tubulin (red) fluorescence nearly fully overlaps.
We stimulated myotube formation in cycling C2C12 myoblasts to test the theory that Acheron binds also to microtubules in differentiated skeletal muscle. DNA (blue), -tubulin, and Acheron were used to stain myotubes (red). Then, using a Nikon A1R-SIMe resonant scanning confocal super resolution microscope, slides were scanned at a magnification of 60X. (Figure 4a). Myoblasts are shown in this figure as mononucleated cells, while myotubes are represented as multi-nucleated cells, demonstrating complete differentiation of each tube. Here, colocalization is shown by the appearance of Acheron in yellow areas of the picture with -tubulin. From Figures 3a and 3c, there is also a noticeable rise in acheron fluorescence (and hence, expression). This supports earlier findings that distinct C2C12 myotubes express more Acheron.
In addition, confirms our theory that in differentiated myotubes, Acheron binds to microtubules (Figure 4a). We once again conducted a visual analysis of the photographs to better clarify these conclusions.
The DAPI signal is shown as the blue line in this graph, whereas Acheron and -tubulin are shown as the green and red lines, respectively, in Figures 3b and 3d (Figure 4b). Our multi-nucleated myotube's numerous nuclei are indicated by a number of blue peaks. Once again, the expression patterns of acheron (green) and -tubulin (red) are essentially identical (Figure 4b). This strengthens our claim that differentiated C2C12 myotubes of Acheron bind microtubules.
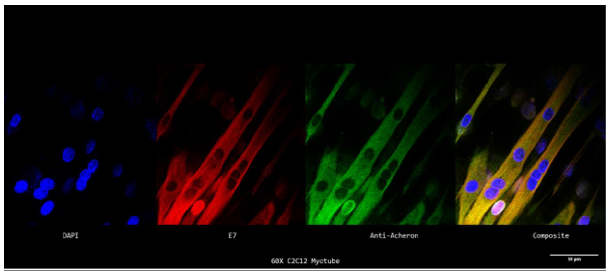
Figure 4a. Super Resolution Subcellular Localization of Acheron Relative to Microtubules in C2C12 Myotubes.
A 60X confocal z-stack of a single labelled cycling mononucleated C2C12 myoblast's nucleus (DAPI; blue; far left), microtubules (E7; red; second to the left), Acheron (anti-Acheron; green; second to the right), and composite (merge; far right) channels.
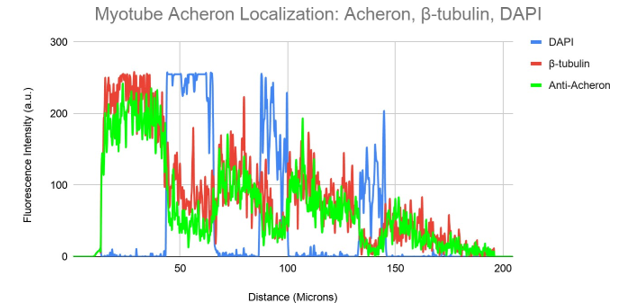
Figure 4b. Colocalization analysis of DAPI, Acheron and β-tubulin Fluorescent Signal From Figure 4a.
DAPI (blue), -tubulin (red), and Anti-Acheron (green) fluorescence intensity is shown on a graph in relation to cell surface area. While the numerous nuclei of the multi-nucleated myotube may be observed as independent DAPI fluorescence peaks, the intensity of the acheron (green) and -tubulin (red) fluorescence significantly overlaps.
We used human U2-OS osteosarcoma cells to investigate the idea that Acheron attaches to microtubules in non-muscle cells. A 15-year-old girl's moderately differentiated sarcoma of the tibia gave rise to the bone cancer cell line known as U2-OS (Rousseau, 2010). These cells' huge size and flat form make them often used in fluorescence microscopy (Rousseau, 2010). High resolution imaging and high accuracy colocalization analyses are made possible by these properties.
Additionally, U2-OS cells are a useful tool for expanding our findings since they endogenously express Acheron (Human Protein Atlas). The same procedures used for C2C12 myoblasts were used to prepare the samples for immunofluorescence, and they were then probed for Acheron (green), DNA (blue), and -tubulin (red). Then, using a Nikon A1R-SIMe resonant scanning confocal super resolution microscope, slides were scanned at a 100X magnification (Figure 5a). In the case of this Figure, the scanning confocal super resolution microscope's resolution and some good timing enabled us to see a cell moving through mitosis in order to collect extra data in addition to testing our theory. As might be predicted, DAPI staining can be detected in the cell's nucleus in Figure 5a, but there are also several distinct foci visible. Condensed chromosomes that are connected to kinetochore microtubules and aligning to the metaphase plate may be seen in this picture thanks to DAPI labelling. On the chromosomes, tension is being applied by the microtubules (red). Microtubules in cycling cells that are in metaphase and anaphase commit themselves to the process of cell division (Oriola et al.,2018). Acheron (green) is predominantly visible colocalizing with the cell's microtubules (yellow) (Figure 5a). The information from this picture, when combined, not only confirms our theory that Acheron attaches to microtubules in many mammalian lineages, but also shows that Acheron/microtubule interaction is maintained even during cell division phases when microtubules are used in different ways, such metaphase (Figure 5a).
We can visually see our colocalization in relation to the cell's size (in microns) by graphic analysis of the picture, and we can also determine the relative expression levels of our examined proteins and structures (Figure 5b). The blue line in this graph is DAPI staining, whereas the green and red lines, respectively, indicate Acheron and -tubulin. Acheron fluorescence can be detected nearly exactly matching the -tubulin fluorescence pattern, similar to Figures 3b, d, and 4b, whereas DAPI signal can be discretely seen. These results may be utilised to show that our yellow colocalization signalling really reflects colocalization rather than being an artefact of two signals layered together.
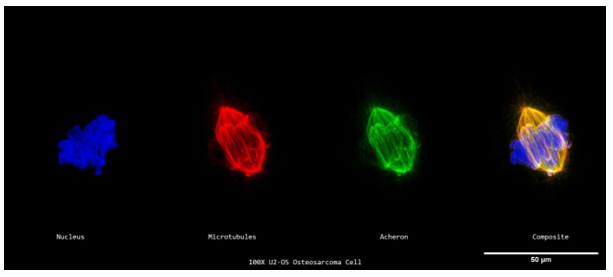
Figure 5a. Acheron’s Microtubule Localization is Preserved Between Different Mammalian Cell Lines.
A 100X confocal z-stack of a single labelled cycling U2-OS osteosarcoma cell's nuclei (DAPI; blue; far left), microtubules (E7; red; second from the left), Acheron (anti-Acheron; green; second from the right), and composite (merge; far right) channels. While chromosomal alignment to the metaphase plate can be identified using the DAPI signal, a significant percentage of the cell's Acheron can be seen colocalizing with microtubules in the yellow portions of the picture.
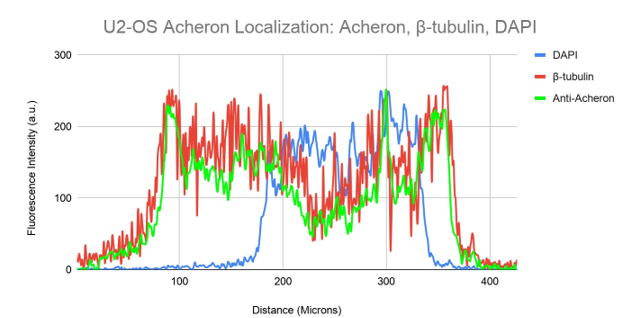
Figure 5b. Colocalization analysis of DAPI, Acheron and β-tubulin Fluorescent Signal From Figure 5a.
An illustration showing the relative fluorescence intensity of DAPI (blue), -tubulin (red), and Anti-Acheron (green) on the cell. While DAPI (blue) peaks can be detected in their own distinctive regions of the cell, Anti-Acheron (green) fluorescence intensity can be seen following the same distal trajectory as -tubulin (red).
We postulated that depolymerizing microtubules would release Acheron into the cytoplasm, allowing us to better assess if Acheron physically attaches to microtubules in mammalian cells. In order to dismantle microtubule connections and promote microtubule depolymerization, we used the anti-cancer drug Nocodazole to test this theory (Verstraelen et al., 2017). After giving Nocodazole to the cells, it was checked to see whether Acheron had localised to the microtubules.
I carried out a dose/response study to assess microtubule depolymerization using immunofluorescence and apoptosis in order to establish the best Nocodazole dosage (phase contrast imaging of membrane blebbing). These research' findings enabled us to identify the ideal Nocodazole treatment parameters that efficiently depolymerized microtubules while preventing cell death. Phase imaging allows for the observation of membrane blebbing and vesicle production at dosages of 750 nM for one hour (Figure 6a). Because dosages over this range would cause cell death, they were immediately disregarded from microtubule analysis. Nocodazole was diluted below 750 nM for an hour before cells were stained for microtubules (E7; magenta) and nuclei (DAPI; cyan), which were then photographed at 60X. (Figure 6b). Until 400 nM for 1 hour, nocodazole treatment failed to depolymerize microtubules in C2C12 myoblasts. Microtubules seem to have totally depolymerized at this time (Figure 6b). Together, the results of these investigations show that 400–750 nM for an hour is the optimal concentration and duration for Nocodazole-mediated microtubule depolymerization in cycling C2C12 myoblasts.
After that, 500 nM Nocodazole was applied to C2C12 cells for 1 hour. The cells were then fixed on poly-L-lysine-treated coverslips and probed for Acheron (green), DNA (blue), and -tubulin (red). Then, using a Nikon A1R-SIMe resonant scanning confocal super resolution microscope, slides were scanned at a 100X magnification (Figure 6c). Microtubule pieces may be observed in red in this illustration, which shows that the microtubule has completely depolymerized. However, in regions of yellow, Acheron (green) may still be observed localising to -tubulin (red) (Figure 6c). Perinuclear regions of the cell exhibit a buildup of Acheron and -tubulin, but the lack of membrane blebbing and vesicle formation indicates that the 500nM Nocodazole treatment did not cause the cell to undergo apoptosis (Figure 6c).
This data is visually displayed in Figure 6d. Acheron (green) and -tubulin (red) peaking may be detected in regions close to the DAPI (blue) signal, which indicates perinuclear accumulation.
Additionally, Acheron expression may be shown to follow a path similar to that of -tubulin, with decreased expression of Acheron in the cell's periphery (Figure 6d). These results confirm our earlier research, which suggests that Acheron binds to individual -tubulin monomers as well as microtubules (Figure 6c, Figure 6d).
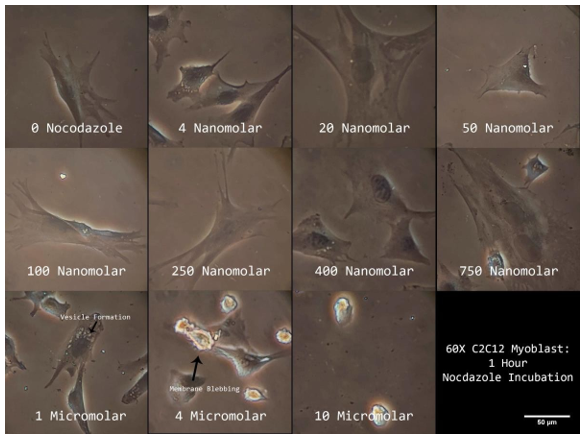
Figure 6a. Phase Contrast Images of Nocodazole Treated C2C12 Myoblasts.
Cycling mononucleated C2C12 myoblasts' phase z-projections in response to various Nocodazole doses. Arrows point to regions that exhibit signs of apoptosis such vesicle production and membrane blebbing. imaged 60 times.
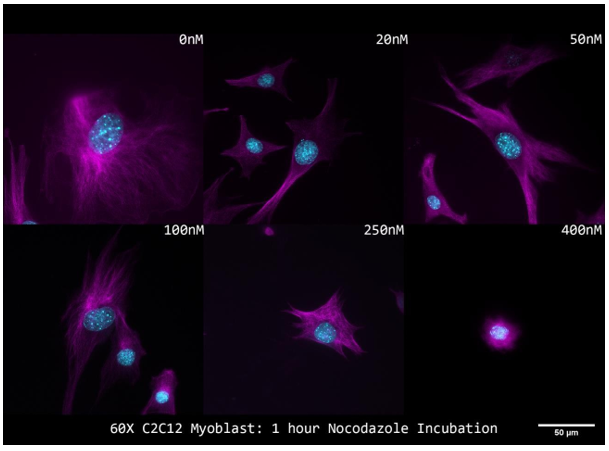
Figure 6b. Immunofluorescent Staining of Nocodazole Treated C2C12 Myoblasts.
Composite z-projections of microtubules (E7; magenta) and nuclei (DAPI; cyan) in cycling mononucleated C2C12 myoblasts in response to different Nocodazole doses. imaged 60 times.
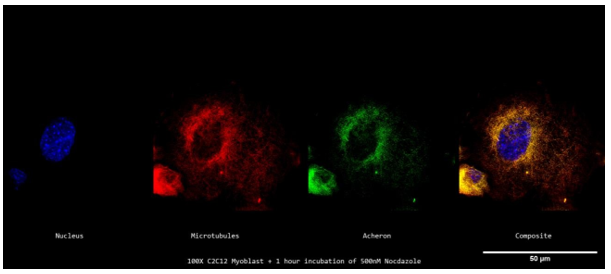
Figure 6c. Acheron localizes to Individual β-tubulin Monomers in Nocodazole Treated C2C12 Myoblasts
A confocal z-stack of a single stained cycling mononucleated C2C12 myoblast treated with 500 nM Nocodazole for an hour, imaged at 100X, showing the nucleus (DAPI; blue; far left), microtubules (E7; red; second to the left), Acheron (anti-Acheron; green; second to the right), and composite (merge; far right) channels. Even though the bulk of the Acheron signal is peri-nuclear in this image, it is still possible to observe Acheron colocalizing with microtubules.

Figure 6d. Colocalization analysis of DAPI, Acheron and β-tubulin Fluorescent Signal From figure 6c.
DAPI (blue), -tubulin (red), and Anti-Acheron (green) fluorescence intensity is shown on a graph in relation to cell surface area. Despite microtubule depolymerization, DAPI signal can only be observed in the nucleus whereas Acheron can be seen in the same regions as depolymerized tubulin monomers.
BAD, HHARI and CASK Localization in C2C12 Myoblasts:
Acheron is essential to PCD, myogenesis, and pathogenesis processes. The BH3-only protein BAD, the E3 ubiquitin ligase HHARI, and the calcium/calmodulin-dependent serine protein kinase, CASK, are thought to interact with Acheron. Therefore, knowing how these chemicals are localised inside the cell in relation to Acheron and figuring out if these molecules are directly complexed with Acheron would be crucial knowledge about Acheron's function in these cellular processes. C2C12 myoblasts were plated on poly-L-lysine coverslips and immunostained for BAD, HHARI, and CASK in relation to microtubules in order to test the hypothesis that Acheron co-localizes with known binding partners (BAD, HHARI, and CASK) at the cell's microtubules. As stated before orange/yellow signals for the other photos may be used to estimate regions of colocalization between BAD, HHARI, CASK (green), and -tubulin (red).
In C2C12 myoblasts, co-IP of Acheron revealed a physical interaction with the BH3-only protein BAD (Sheel et al., 2020). I wanted to find out whether BAD could be discovered localising to microtubules in C2C12 myoblasts given our earlier discoveries that Acheron attaches to microtubules and that BAD could be found binding to Acheron. Stains for DNA (blue), BAD (green), and -tubulin were applied to myoblasts (red). Then, using a Nikon A1R-SIMe resonant scanning confocal super resolution microscope, slides were scanned at a 100X magnification (Figure 7a). The absence of yellow regions in the final composite picture suggests that there is no detectable colocalization. Instead of the filamentous pattern that is typical of Acheron, BAD (green) may be observed following a punctate pattern (Figures 3a; 3c; 4a; and 5a).
The fluorescence intensities of each colour channel are shown graphically to support our conclusion that BAD does not bind to microtubules in C2C12 myoblasts (Figure 7b). Here, the BAD (green) signal exhibits a fluorescence pattern distinct from that of -tubulin (red) and DAPI (blue). Similar to our previous expression of mitotracker, peaks of BAD expression may be found in perinuclear regions of the cell (Figures 1a-c). These findings do not, taken together, support the idea that BAD localises to microtubules in C2C12 myoblasts.
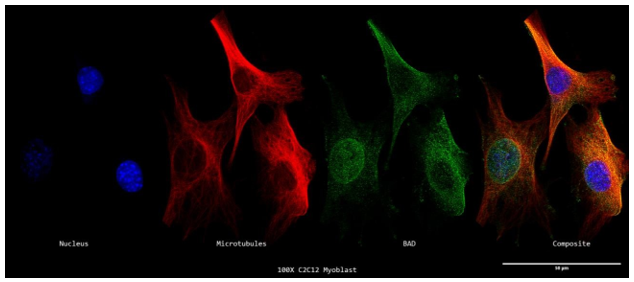
7a. Super Resolution Subcellular Localization of BAD Relative to Microtubules in C2C12 Myoblasts.
A 100X confocal z-stack of a single labelled cycling C2C12 myoblast's nuclei (DAPI; blue; far left), microtubules (-tubulin; red; second from the left), BAD (anti-BAD; green; second from the right), and composite (merge; far right) channels.
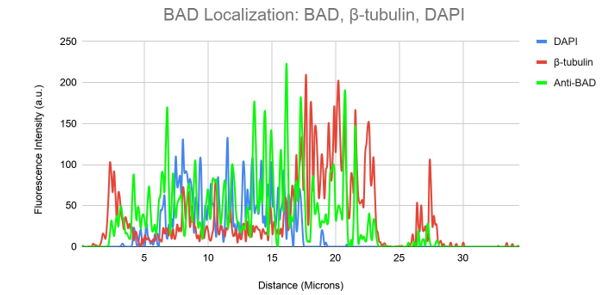
Figure 7b. Colocalization analysis of DAPI, BAD and β-tubulin Fluorescent Signal From Figure 7a.
DAPI (blue), -tubulin (red), and Anti-BAD (green) fluorescence intensity is shown on a graph in relation to cell surface area. While partially overlapping with the DAPI signal, the anti-BAD (green) fluorescence intensity can be observed travelling in a different distal direction from the -tubulin (red).
Acheron and Human Homolog of Ariadne-1 (HHARI), a ubiquitin ligase, were shown to be bound by Co-IP and Yeast 2-Hybrid screening (Wang Z, 2003). This information, together with the knowledge that Acheron attaches to microtubules in mammalian cells, prompted me to investigate the possibility that HHARI may also be connected to microtubules in mammalian cells.
DNA, HHARI, and -tubulin were all labelled on C2C12 cells in blue, green, and yellow (red). Then, using a Nikon A1R-SIMe resonant scanning confocal super resolution microscope, slides were scanned at a 100X magnification (Figure 8a). Similar to Figure 7a, there is no discernible area of yellow staining, indicating that colocalization is not present. Instead, HHARI is shown to collect in certain perinuclear regions of the cell. It may be assumed that HHARI is localising to the golgi apparatus, with some extra signal in the nucleoplasm, rather than being at the cell's centrosome since this region does not colocalize with microtubules (Figure 8a).
Figure 8b illustrates this in a visual way. The HHARI fluorescent signal is shown in green in this figure, whereas the -tubulin and DAPI signals are shown in red and blue, respectively. Here, HHARI seems to express independently of -tubulin, accumulating at one end of the nucleus and exhibiting extra expression inside the cell nucleus (Figure 8b).
The conclusion that HHARI attaches to microtubules in C2C12 myoblasts is not supported by our results.
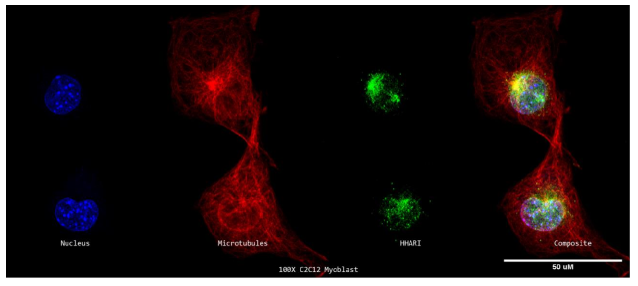
8a. Super Resolution Subcellular Localization of HHARI Relative to Microtubules in C2C12 Myoblasts
A 100X confocal z-stack of a single labelled cycling C2C12 myoblast's nuclei (DAPI; blue; far left), microtubules (-tubulin; red; second from the left), HHARI (anti-HHARI; green; second from the right), and composite (merge; far right) channels.
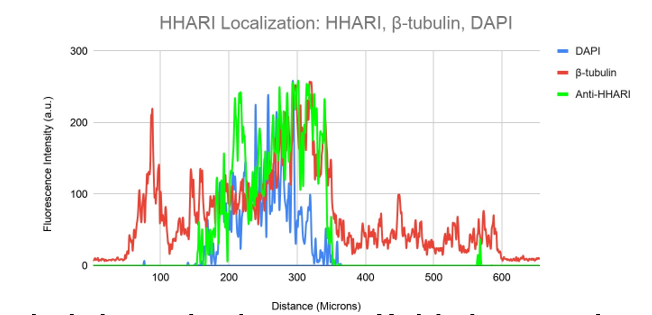
Figure 8b. Colocalization analysis of DAPI, HHARI and β-tubulin Fluorescent Signal From Figure 8a.
DAPI (blue), -tubulin (red), and Anti-HHARI (green) fluorescence intensity is shown on a graph in relation to cell surface area. In this image, HHARI fluorescence is seen.
There are significant quantities outside the nucleus and minimal amounts within. The signals here do not match the fluorescence of -tubulin.
Acheron and mouse embryonic cDNA were subjected to yeast 2-hybrid screening, which demonstrated CASK interacts to the C-terminus of Acheron through CASK's CaM K II-like domain (Wang et al., Unpublished).
It is generally known that CASK is localised to the cytoplasm and nucleus of the cell. We hypothesised that some of CASK may localise to regions of known Acheron localisation in mammalian cells as a result of our discovery that CASK binds to Acheron, a protein that is localised to microtubules.
C2C12 myoblasts were stained for DNA (blue), CASK (green), and -tubulin to test the theory that CASK localises to microtubules of C2C12 cells (red). Then, using a Nikon A1R-SIMe resonant scanning confocal super resolution microscope, slides were scanned at a 100X magnification (Figure 9a). Similar to Figures 7a and 8a, this picture has no yellow coloration at all, suggesting that colocalization is not present. In accordance with data reported in the literature (Human Protein Atlas), CASK localises to the cytoplasm and nucleus in this figure (Figure 9a). Figure 9b makes this extremely clear. With the exception of a minor spike in CASK signal that overlaps with DAPI, which indicates that some CASK localises to the nucleus, CASK (green), tubulin (red), and DAPI (blue) signal seem to occupy separate sites inside the cell in the image (Figure 9b). These findings corroborate the idea that CASK localises to the nucleus, a region that is often linked to Acheron localization.
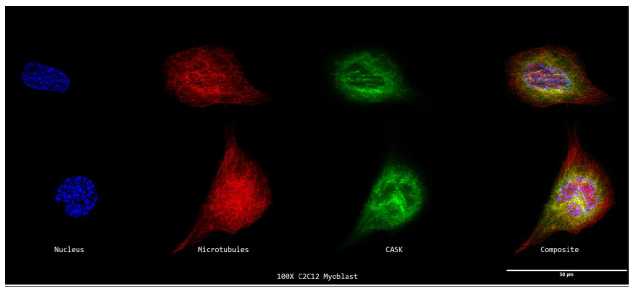
9a. Super Resolution Subcellular Localization of CASK Relative to Microtubules in C2C12 Myoblasts
A 100X confocal z-stack picture showing the cycling C2C12 Myoblasts' nuclei (DAPI; blue; far left), microtubules (E7; red; second from the left), CASK (anti-CASK; green; second from the right), and composite (merge; far right) channels.
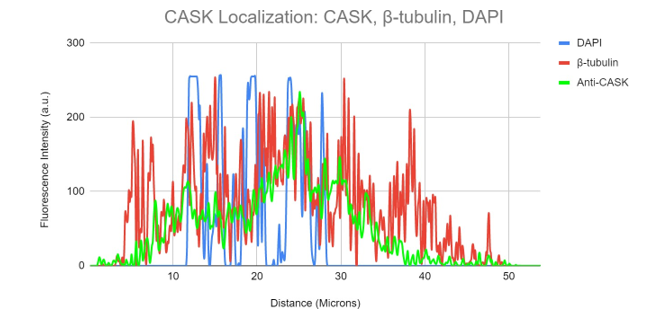
Figure 9b. Colocalization analysis of DAPI, CASK and β-tubulin Fluorescent Signal From Figure 9a.
DAPI (blue), -tubulin (red), and anti-CASK (green) fluorescence intensity is shown on a graph in relation to cell surface area. Anti-CASK (green) fluorescence intensity can be observed in this figure travelling in a different distal route from -tubulin (red).
Anti-CASK expression may be detected accumulating in the nucleus close to the DAPI signal.
Nuclear export signals (NES) and functional nuclear localization signals (NLS) are features of the dynamic protein acheron (Valavanis et al., 2009; Shao et al., 2012). According to preliminary research, Acheron localises to the nucleus as well as microtubules (Fig. 1c). Growing and waning levels of growth factors, respectively, may cause Acheron to translocate into and out of the nucleus (Glenn et al., unpublished). Although CASK's cytoplasmic localization in the cytoplasm and nucleus of the cell has been well established (Gardener et al., 2006), CASK lacks a nuclear localization signal, hence the mechanism that promotes translocation is not entirely evident. According to earlier research, CASK is nuclear translocated as a consequence of ectopic expression of T-box transcription factor T-brain-1 (Tbr-1), which is situated in the cytoskeletal structure (Bredt et al., 2000). We wondered whether Acheron served as a shuttle protein for CASK entering the nucleus in light of these results and the knowledge that it binds to CASK and localises to microtubules.
We used two distinct lines of C2C12 cells producing ectopic Acheron to test the idea that CASK translocates to the nucleus when Acheron translocation is stimulated (Shao et al., 2012). A plasmid encoding the expression of full-length, HA-tagged Acheron, driven by a pCMV-Neo retroviral promoter, was transfected into the first line. A mutant Acheron that lacks the nuclear export signal was transfected into the second line (Shao et al., 2012). We can see the cells in a time-independent manner as a result of nuclear Acheron being locked in the nucleus.
Both lines were plated on coverslips coated with poly-L-lysine and developed in conventional C2C12 growth media. Cells were fixed when Acheron was lured into the nucleus by increasing growth factor concentrations as previously mentioned after they had attained 70% confluency. Then, cells were photographed at 100X while labelled for Acheron (green), -tubulin (red), and DAPI (blue) (Figure).
10a). For this experiment, the cell lines C2C12 + HA + Acheron and C2C12 + HA + Acheron -NES were used, and control pictures of both cell lines are shown in additional Figures 2a and 3a. Acheron is seen in these photos confined to the cell's microtubules. Acheron doesn't start to collect around the nucleus in NES cells until serum levels are high.
Figure 10a shows cells cultivated in conventional 10% FBS in the top half and cells cultured in 15% FBS with nonfunctional NES in the bottom half. In line with our earlier photos, acheron (green) can be shown confined to microtubules (red). The bright yellow colour in the composite picture on the right serves as evidence of this (Figure 10a). However, Acheron (green) got stuck in the nucleus (blue) and had reduced levels of expression linked to microtubules (red) when -NES cells were incubated in 15% FBS (Figure 10a).
Figures 10b and 10c show this in considerably more depth. The 10% FBS sample's fluorescence intensity are shown visually in Figure 10b. Following the expression of -tubulin (red), the Acheron fluorescence intensity (green) may be detected in this image. Low amounts of Acheron staining may be found in the nucleus close to the DAPI (blue) signal (Figure 10b).
Figure 10c, on the other hand, displays the fluorescence intensity of the 15% -NES sample. In this picture, Following the -tubulin's (red) fluorescence intensity, although to a lesser degree, is the expression of acheron (green). Instead, near the cell nucleus, sizable peaks of the Acheron (green) signal can be observed aggregating with the DAPI (blue) signal. Together, the findings show that Acheron may be pushed into the cell's nucleus in response to rising growth factor concentrations.
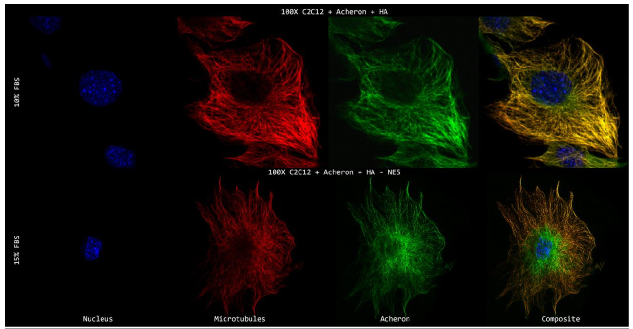
Figure 10a. Acheron Nuclear Translocation in Response to Increased Growth Factors in C2C12 Myoblasts.
A 100X confocal z-stack of a single labelled modified C2C12 cell showing the nuclei (DAPI; blue; far left), microtubules (E7; red; second from the left), Acheron (anti-Acheron; green; second from the right), and composite (merge; far right) channels.
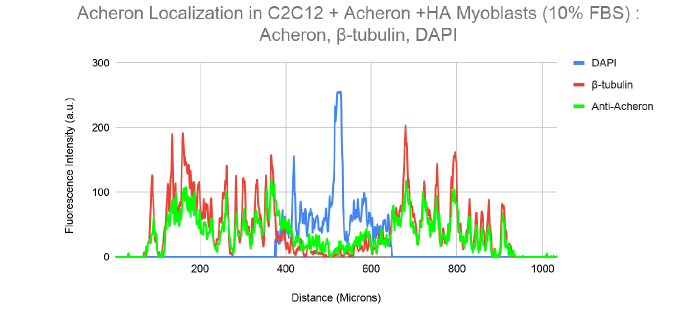
Figure 10b. Colocalization analysis of DAPI, Acheron and β-tubulin Fluorescent Signal in C2C12 Myoblasts in 10% FBS.
An illustration showing the relative fluorescence intensity of DAPI (blue), -tubulin (red), and anti-Acheron (green) in 10% FBS samples. In this figure, the expression location for the -tubulin (red) signal matches the anti-Acheron (green) fluorescence intensity. DAPI (blue) signal is close to low levels of Achron (green) in the nucleus.
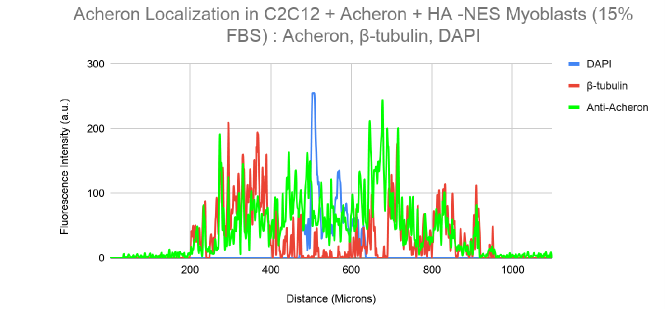
Figure 10c. Colocalization analysis of DAPI, Acheron and β-tubulin Fluorescent Signal in C2C12 Myoblasts in 15% FBS.
An illustration showing the relative fluorescence intensity of DAPI (blue), -tubulin (red), and anti-Acheron (green) in samples of 15% FBS. Unlike Picture 10b, which shows Acheron (green) aggregating around DAPI (blue) signal in the cell nucleus, this figure shows anti-Acheron (green) fluorescence intensity matching expression localisation for -tubulin (red) signal.
The experiment was repeated with the exception that slides were probed for CASK (green), -tubulin (red), and DNA (blue) in order to further investigate the idea that CASK translocates to the nucleus of mammalian cells when Acheron translocation is stimulated (Figure 11a).
The panel of photographs that results is structured similarly to Figure 10a. Cells that were not exposed to higher growth factor concentrations can be seen in the top half of the picture, whereas NES cells that were incubated in higher serum concentrations can be seen in the bottom half.
CASK (green), which is distinct from microtubules, is confined to the cytoplasm of the cell in the 10% FBS cells (red). This is consistent with the findings in Figure 9a. Different behaviour of CASK (green) is seen in the 15% FBS-NES cells. Contrary to earlier, there is a large rise in the expression of CASK (green) in these cell lines, and the green signal is far more common than its 10% FBS counterpart. Some of the signal localises to microtubules (red), as shown by the yellow colouring, with the majority of the signal also being visible in the nucleus (blue) (Figure 11a). In Figures 11b and 11c, the localisation of CASK in both samples is more clearly shown visually.
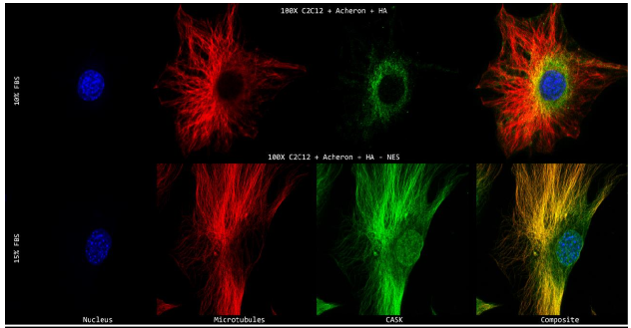
Figure 11b depicts the perinuclear space, where CASK fluorescence intensities (green) can be observed accumulating surrounding the DAPI (blue) signal. Additionally, the CASK (green) signal exhibits a fluorescence intensity pattern distinct from the -tubulin (red) signal (Figure 11b).
Figure 11c depicts the accumulation of CASK fluorescent signal (green) in the nucleus near DAPI (blue) signal to a larger degree than Figure 11b. Comparatively, this shows that CASK (green) successfully moved to the cell's nucleus in response to rising growth stimuli. The CASK (green) fluorescent signal can be observed closely following the -tubulin (red) signal in 15% FBS samples, in sharp contrast to our earlier CASK findings, demonstrating localization to microtubules (Figure 11c).
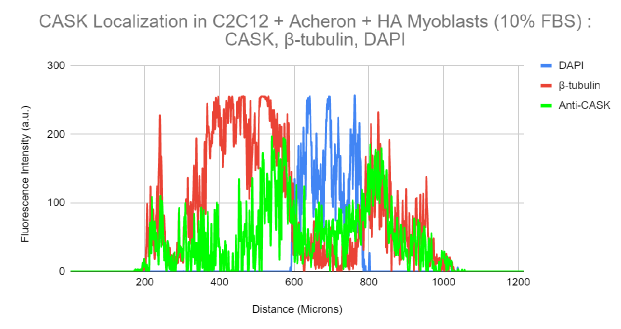
Figure 11a. CASK Nuclear Translocation in Response to Increased Growth Factors in C2C12 Myoblasts.
A 100X confocal z-stack of a single stained modified C2C12 cell showing the nuclei (DAPI; blue; far left), microtubules (E7; red; second from the left), CASK (anti-CASK; green; second from the right), and composite (merge; far right) channels.
.png)
Figure 11b. Colocalization analysis of DAPI, CASK and β-tubulin Fluorescent Signal in C2C12 Myoblasts in 10% FBS.
An illustration showing the relative fluorescence intensities of DAPI (blue), -tubulin (red), and Anti-CASK (green) in 10% FBS samples. In this image, anti-CASK (green) fluorescence can be seen adhering to -tubulin (red) along the fluorescent signal intensity channel and accumulating towards the DAPI (blue) signal in the perinuclear region.
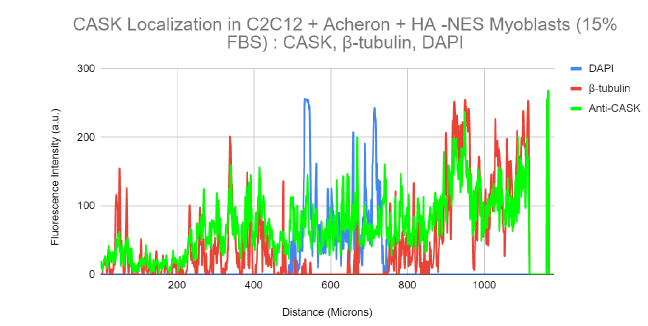
Figure 11c. Colocalization analysis of DAPI, CASK and β-tubulin Fluorescent Signal in C2C12 Myoblasts in 15% FBS.
An illustration showing the relative fluorescence intensity of DAPI (blue), -tubulin (red), and Anti-CASK (green) in samples of 15% FBS. As compared to its 10% FBS counterpart, the anti-CASK (green) fluorescence intensity in this figure can be observed localising to the DAPI (blue) signal in higher amounts. In striking contrast to Figure 11b, the intensity of the CASK (green) fluorescence signal can be observed closely following the expression of -tubulin (red) as well.
Discussion:
The trials mentioned above have two different investigations as their goals. In the first investigation, I aimed to identify Acheron's subcellular location. I investigated the theories that Acheron localises to mitochondria, binds to actin, and binds to microtubules in mammalian cells in order to achieve this objective. I used developed C2C12 myotubes, U2-OS osteosarcoma cells, and undifferentiated C2C12 myoblasts. The idea that Acheron localization was taking place directly via interactions with tubulin was subsequently put to the test.
In the second experiment, I investigated the possibility that Acheron colocalizes with a set of known binding partners, including BAD, Human Homology of Ariadne-1 (HHARI), and Calcium/Calmodulin-dependent Serine Protein Kinase (CASK). I further explored the idea that Acheron may aid in the movement of CASK into mammalian cells' nuclei.
Overall, these research' findings indicate that Acheron binds to microtubules in several cell types and that this localisation is directly controlled by Acheron—tubulin binding. These research' findings, however, did not corroborate the idea that Acheron and BAD and Acheron and HHARI colocalize at the cell's microtubules. The results of this investigation did support the idea that Acheron could make it easier for CASK to enter mammalian cells' nuclei.
Figures 1-6 show the results of the first investigation. Acheron was mostly visible in the nucleus and followed a reticulated pattern in the cytoplasm in Figure 1a. However, the cytoplasmic signal had a particular reticulated pattern (Figure 1a). Graphically, the Acheron signal can be shown occupying area that is essentially independent of the Mitotracker fluorescence, despite the Acheron and Mitotracker signals' proximity to the nucleus (45 um and 90 um). Although not with the maxima of the DAPI (blue) signal detected about 55um and 75um, acheron fluorescence did seem to overlap with the DAPI signal in the nucleus. Figure 1a shows these peaks as the brighter blue patches that may be densely packed heterochromatin in the cell's nucleus (Linhoff et al., 2015). Although further direct testing is needed, the separation of Acheron fluorescence and heterochromatin supports the theories that, although being present in the nucleus, Acheron does not attach to the DNA directly. Acheron signal in the cytoplasm seen in Figure 1 is not diffuse, but rather follows a particular cytoskeletal structure, as can be seen by closer inspection. This prompted us to postulate that Acheron binds to a component of the cytoskeleton like actin or tubulin. Nuclear Acheron looks diffuse rather than following a recognisable pattern, in contrast to its cytoplasmic staining. It's interesting to note that nuclear and cytoskeletal signals are not necessary for perinuclear Acheron to seem to follow its own distinct pattern. A distinct virtually horizontal pattern can be seen projecting out of the nucleus into the cytoplasm, whereas Acheron can be seen around the nuclear membrane in Figure 1c's improved resolution. This prompts us to speculate that Acheron may be localising to ribosomes and the cell's rough endoplasmic reticulum, respectively.
Aggregates of Acheron were seen near the cell's perimeter, correlating with the results provided by Dermit et al. (2020). (Figure 1b). As would be predicted for lamellipodia, these regions of increased Acheron signal do not localise to the leading edge of the cell. Instead, these regions of increased Acheron signal localise to filopodia-like protrusions that protrude from the cell. This lends credence to recent research by Dermit et al. (2020) showing that Acheron is abundant at actin-rich cell protrusions. When combined, the findings from Figures 1a, 1b, and 1c lend credence to the idea that Acheron attaches to a cytoskeletal component there. Additionally, the data collected for this research gives us better resolution and details on Acheron's localization both inside the nucleus and in its enriched regions at the cell's filopodia.
But examination of the Acheron staining in this cell confirms many of the findings from Figures 1a, b, and c. A pattern seen in the nucleus of acheron is expressed more strongly in the perinuclear region. Acheron expression exhibits a reticulated pattern in the cytoplasm, according to a closer inspection (Figures 1a and 1c). As previously, brighter regions of enhanced Acheron signal are seen at the ends of actin filaments. This bolsters the claims that: a) the Acheron staining zones are real and consistent with the results provided by Dermit et al. (2020); and b) the enhanced areas may be detected at the cell's filopodia rather than merely at cellular protrusions. Acheron does not localise to actin filaments, despite the fact that filopodia are actin-rich structures, as shown by the information above (Figures 2a and 2b). We examined cells for microtubules, a distinctive cytoskeletal component linked to filopodia, in an effort to solve this puzzle. Figures 3a and 3c show the results of our Acheron, DNA, and microtubule probes on cycling C2C12 myoblasts. In contrast to Figures 1 and 2, the cytoplasmic Acheron and microtubule fluorescence signals are colocalized in these photos, to the best of their ability. Fluorescence in the yellow colour indicates this (Figures 3a, 3c). The idea that Acheron localises to microtubules in mammalian cell lines is supported by this.
We differentiated C2C12 myoblasts into myotubes and stained them for Acheron, DNA, and microtubule filaments to test the theory that Acheron-microtubule localization is maintained with throughout development (Figure 4a). In the ensuing image, we discovered that Acheron expression had been significantly elevated and that, in contrast to the predominant nuclear staining observed in cycling cells, the signal was mostly cytoplasmic and linked to the cytoskeleton. Figure 4a shows this visually, and Figure 4b shows it graphically. When DAPI expression increases, Acheron expression dips, indicating that in differentiated myotubes, Acheron has a preference for the cytoplasm rather than the nucleus (Figure 4b). This implies that in mouse myoblasts and myotubes, acheron location is developmentally controlled. This leads us to hypothesise that Acheron is limited to the cytoplasm after differentiation and has functions in the nucleus and cytoplasm of cycling cells. (See Figure 4a.)
We immunostained cycling U2OS human osteosarcoma cells for Acheron, DNA, and microtubules in order to further our investigations and ascertain if Acheron binding to microtubules is exclusive to C2C12 cells or a more widespread phenomena (Figure 5a). In U2OS cells, Acheron binds to microtubules, and this was verified. The yellow fluorescence in the composite channel and the overlap between the graphed Acheron (green) and -tubulin (red) fluorescence intensities serve as examples of this (Figure 5a and 5b). We also see nuclear Acheron staining (green), which appears to be excluded from the DNA DAPI (blue) signal, which is consistent with the findings from C2C12 cells (Figure 5b). We had the good fortune to photograph a cell during the metaphase of mitosis for this figure. As they align to the metaphase plate, the condensed chromosomes may be observed connected to kinetochore microtubules. Microtubules and Acheron co-localize, but not chromosomes (Figure 5a and 5b). Considering all of the information, it is suggested that Acheron attaches to microtubules and that this Acheron-microtubule interaction is conserved in mammalian species. Additionally, it supports the idea that nuclear Acheron does not directly attach to DNA.
Finally, cycling C2C12 myoblasts were treated with Nocodazole and probed for Acheron, DNA, and microtubules in order to test the idea that Acheron-microtubule localization is preserved when susceptible to microtubule depolymerization (Figure 6c). In this case, 500 nM of nocodazole for an hour was shown to be the best dosage. It was discovered that although this dose completely depolymerizes microtubules, it does not instantly cause apoptosis (Figures 6a and 6b). Surprisingly, Acheron was discovered to maintain microtubule binding even after depolarization, as seen by yellow fluorescence in the composite channel. Although the acheron signal is obviously diffuse, it is colocalized with -tubulin monomers as seen by the yellow fluorescence and signal overlap that can be detected (Figure 6c and 6d). This offers fresh understanding of Acheron's microtubule localisation. In light of the aforementioned information, the results of this test are consistent with the idea that Acheron localises to microtubules in mammalian cells and that this localization is unique to the monomer of the microtubule, -tubulin (Figure 6c and 6d).
Acheron's La theme lends more weight to this assertion. A functional nuclear localization signal (NLS), a nuclear export signal (NES), a Suz-C domain (subcellular localization and mRNA substrate recognition), a well-conserved RNA recognition motif (RRM), and a functional, winged helix-turn-helix structured RNA binding, LA motif (LAM) are all present in Acheron's structure (Stefanovic et al., 2014; Martino et al., 2015; Bousquet-Antonelli et al., 2009; Dong et al., 2004; Valavanis et al., 2007; Stavraka et al., 2015). Individual alpha and beta tubulin monomers interact with winged-helix protein domains through its carboxy-terminus, according to X-ray crystallography and crosslinking/mass spectrometry (MS) findings (Abad et al., 2014).
The localization of Acheron in the nucleus, perinuclear, and filopodia, as well as other aspects of Acheron localization, are all revealed by Acheron staining under diverse experimental settings. Acheron's perinuclear expression is further clarified by the enhanced yellow fluorescence in the nucleus. While this increase in Acheron expression could indicate that the protein localises to the cell's Golgi apparatus or rough endoplasmic reticulum, the presence of yellow colocalization with microtubules tells us that this localization is more likely to the centrosome, one of the main microtubule organising centres of the cell. Filopodia have a lot of actin (Mattila and Lappalainen, 2008). We discovered that Acheron is concentrated at the filopodia but does not bind with phalloidin-stained structures inside the cell (Figure 2). (Figure 2b). The role of microtubules in cell proliferation provides the best explanation for this occurrence. Microtubules and actin filaments compete with one another to control cell mobility inside growth cone-like structures during cell proliferation and migration (Khan and Baas, 2016). Growth cones' main structures are filopodia (Davenport et al., 1993). Acheron may then be restricted to the microtubules in these formations.
The theory that microtubules may directly engage in directed cell motility via filopodia merging and modulation of filopodia density is supported by recent findings as well (Schober et al., 2007).
As a result, one hypothesis is that Acheron may likewise be abundant and localised at the microtubules that regulate the movements of filopodia.
The nucleus exhibits relative levels of the Acheron signal in practically all experimental circumstances. However, the peaks of brighter DAPI expression never correspond with the graphed expression intensities of nuclear Acheron (green) (blue). The information presented in this study suggests that, although Acheron localises to the nucleus, it does not bind directly to DNA and, as a result, does not become enriched at bundles of heterochromatin or condensed chromosomes, contrary to previous studies that have shown pericentric heterochromatin is easily detectable as large DAPI-dense foci (Imai et al., 2017). This sheds fresh light on the mechanism controlling Acheron's RNA binding protein function.
Numerous mRNA molecules reside in the cell's nucleoli. Numerous RNA-binding proteins may also be discovered localised in nucleoli as a result (Nazer et al., 2011). Recent information, however, has highlighted the significance of heterochromatin localization to the cell's nucleoli's periphery. Super resolution microscopy's increased resolution reveals that Acheron does not localise to DNA, and consequently heterochromatin itself, and that Acheron is not present in or around nucleoli (Guetg and Santoro, 2012). This information, along with the fact that Acheron has been shown to play a role in the biogenesis of ribosomes (Dermit et al., 2020), a process that starts in the nucleolus and ends in the cytoplasm of eukaryotic cells, leads us to conclude that Acheron's role in ribosome biogenesis is independent of nucleoli localization and must take place in cytoplasm (Pelletier et al., 2018).
Ribosome biogenesis begins in the nucleolus, where pre-ribosome assembly (pre-40s, pre-60s) takes place. Pre-ribosomes that have been synthesised then go from the nucleoplasm into the cytoplasm, where they develop into the ribosomal 40S and 60S subunits that make up the ribosome (Thomson et al., 2013). When combined, the results of these investigations imply that Acheron controls ribosome biogenesis by using ribosomal subunits. Future research is needed to look at this, however.
In conclusion, the research's studies show that Acheron is abundant in the growth cones of the filopodia and localises to microtubules in mammalian cells, particularly to individual -tubulin monomers. Finally, the evidence points to the possibility that nuclear Acheron may not directly attach to DNA; however, further research is needed to confirm this.
In C2C12 myoblasts, co-IP of Acheron revealed a physical interaction with the BH3-only protein BAD (Sheel et al., 2020). I wanted to find out whether BAD could also be seen localising to microtubules in C2C12 myoblasts given our previous discoveries that Acheron binds to both microtubules and BAD. The findings of this research indicate that BAD does not reside on microtubules (Figure 7). Super resolution microscopy does not support the idea that Acheron and BAD colocalize at the cell's microtubules together, despite co-IP results showing a physical contact between Acheron and the BH3-only protein BAD in C2C12 myoblasts. Post-translational changes of BAD are one theory that might be used.
Five serines in the structure of BAD may be phosphorylated by various kinases (Burlacu et al., 2003). Ser112, Ser128, Ser136, Ser155, and Ser179 are some of these locations. BAD Ser128 may be detected localised to the mitochondria, according to immunofluorescent examination (Yang et al., 2008). BAD, however, is phosphorylated at Ser155, which causes it to go into the cytoplasm and prevents it from attaching to Bcl-2 (Datta et al., 2000). Future studies looking at BAD/Acheron co-localization should keep an eye on where BAD is located in relation to its level of phosphorylation. Phosphorylation often causes conformational changes, which expose residues for several possible binding partners (Groban et al., 2006). We may expand our co-IP data by tracking the localisation of BAD in different phosphorylated states. It was discovered by Co-IP and yeast 2-Hybrid screening that Acheron interacts to the human homolog of Ariadne-1, a ubiquitin E3 ligase (HHARI). Cycling C2C12 myoblasts were immunostained for HHARI, tubulin, and DNA to test the theory that HHARI may be detected localised to microtubules in mammalian cells (Figure 8). HHARI did not localise to the microtubules, as was discovered. This information contradicts earlier results from Co-IP and yeast 2-Hybrid showing HHARI interacts to the microtubule-binding protein Acheron. The peri-nuclear signal from Acheron and HHARI might provide one reason for this disparity.
HHARI does not localise to microtubules, as we have shown. However, the region surrounding the nucleus is rich in both HHARI and Acheron (Figures 3b & 8b). Microtubule-organizing centres (MTOCs) like the centrosome are crucial for microtubule organisation. Microtubule organisation, nucleation, and attachment of microtubule minus ends occur at these locations (Conduit et al., 2015). According to recent research, the Golgi apparatus is another location where microtubules may form and become stable (Chabin-Brion et al., 2001).
As a result, polarised microtubules that originate from the Golgi may encourage cell asymmetry and the polarised transport of post-Golgi carriers necessary for cell migration (Vinogradova et al., 2012). Given that HHARI does not localise to microtubules, it may be assumed that HHARI is localising to the Golgi apparatus rather than centrosomal localization as with Acheron from the rise in perinuclear fluorescence (Figure 8).
The additional information reported in this thesis provides support for one possibility explaining the HHARI/Acheron binding seen using co-IP and Yeast-2-Hybrid. Acheron is more abundant near the cell's filopodia, likely in the growth cones of proliferating cells (Dermit et al., 2020). (Figures 1-6). Dynamic growth cone rotation in proliferating cells requires polarised invasion of microtubules into one side of the peripheral domain of the growth cone (Kahn et al., 2016). Given that the Golgi apparatus functions as an MTOC of polarised microtubules, it is possible that the localization of HHARI to the Golgi and the enrichment of Acheron in growth cones of proliferating cells are related. Analysis of Acheron enrichment at the filopodia in HHARI-deficient cells has to be done in more research.
Acheron and mouse embryonic cDNA were subjected to yeast 2-hybrid screening, which demonstrated CASK interacts to the C-terminus of Acheron through CASK's CaM K II-like domain (Wang et al., unpublished).
It is already well known that CASK localises to the cytoplasm and nucleus of the cell (Ojeh et al., 2008). Acheron localises to the cell's microtubules and nucleus, according to earlier research. I stained cycling C2C12 myoblasts for CASK (green), -tubulin (red), and DAPI to test the idea that CASK localises to the microtubules of mammalian cells (blue). According to the results of this study, CASK does not bind to microtubules in mammalian cells.
Acheron and CASK are revealed by colocalization studies, but they bind in yeast-2-hybrid experiments. The capacity of Acheron to move into and out of the cell's nucleus is one tenable explanation for this data difference.
A functional nuclear export signal (NES) and nuclear localization signal (NLS) are features of the dynamic protein acheron (Shao et al., 2012). Additionally, earlier research from the lab has shown that Acheron translocation may be initiated in response to both rising and decreasing growth factors, respectively (Glenn et al., unpublished). Although CASK is canonically located in the cytoplasm and nucleus of the cell, it lacks a nuclear localization signal (NLS) (Gardener et al., 2006). Because of this, it is unknown what causes its translocation. One hypothesis is that when Acheron shuttles CASK into the nucleus, a brief period of CASK-Acheron binding takes place.
In Figures 10 and 11, I investigated the claim that, in response to rising growth factors, CASK translocation to the nucleus is linked with Acheron translocation to the nucleus. Together, the findings from this research demonstrate that, in response to rising growth factors, CASK translocation to the nucleus is linked with Acheron translocation to the nucleus. Furthermore, the information shown in Figure 11a is consistent with the theory that CASK colocalizes with Acheron at the cell's microtubules during its translocation (Sonnen et al., 2012). (Figures 1-6).
In conclusion, our observations are consistent with the idea that Acheron is concentrated in the growth cones of the filopodia and localises to microtubules, particularly to individual -tubulin monomers. The nuclear acheron does not attach to DNA directly (future studies are required). On the microtubules of cycling mammalian cells, Acheron, BAD, and HHARI do not colocalize. In response to rising growth factors, CASK translocation to the nucleus is associated with Acheron translocation to the nucleus, and ultimately CASK colocalizes with Acheron at the cell's microtubules during its translocation.
Although noteworthy, these results highlight the need for further research to not only examine the processes governing the localization of Acheron, HHARI, BAD, and CASK in mammalian cells but also to validate the information provided above. To verify the visual information provided by fluorescence microscopy above, appropriate biochemical tests such as fractionation and Western blotting, co-IPs, FRET, and should all be carried out. Future research will examine the distribution of BAD and Acheron in different phosphorylated states in mammalian cell lines, the enrichment of Acheron at the filopodia of HHARI-deficient and wild-type mammalian cells, and the distribution of Acheron and CASK in different cellular compartments in response to increasing growth factors, among other things.
Supplemental Figures:
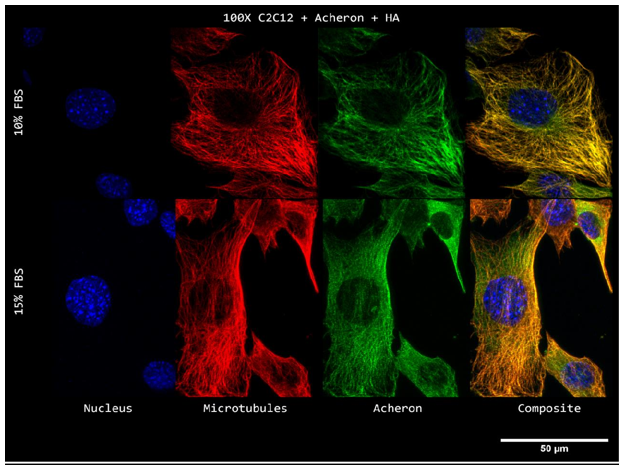
Supplemental Figure 1a. Super Resolution Subcellular Localization of Acheron Relative to Microtubules in C2C12 Myoblasts Without -NES Deletion.
An illustration showing the relative fluorescence intensity of DAPI (blue), -tubulin (red), and Anti-Acheron (green) in samples of 15% FBS.
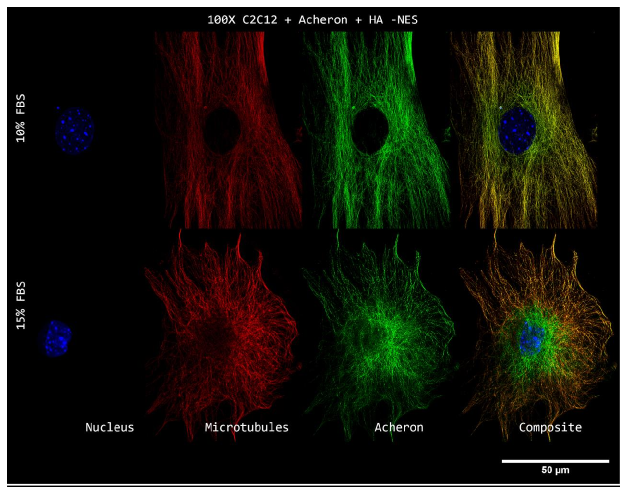
Supplemental Figure 2a. Super Resolution Subcellular Localization of Acheron Relative to Microtubules in -NES C2C12 Myoblasts.
An illustration showing the relative fluorescence intensity of DAPI (blue), -tubulin (red), and Anti-Acheron (green) in samples of 15% FBS. In contrast to its 10% FBS counterpart, anti-CASK (green) fluorescence intensity can be observed localising to DAPI (blue) signal in this figure. In striking contrast to Figure 11b, the intensity of the CASK (green) fluorescence signal can be observed closely following the expression of -tubulin (red) as well.

Table 2a. Appendix of Key Figures and Single Slice Representations:
The file names, cell lines used, staining colour combinations, and single slice representative pictures of the important figures used in this dissertation are included in a supplementary table.
References
Abad, M. A., Medina, B., Santamaria, A., Zou, J., Plasberg-Hill, C., Madhumalar, A., Jayachandran, U., Redli, P. M., Rappsilber, J., Nigg, E. A., & Jeyaprakash, A. A. (2014). Structural basis for microtubule recognition by the human kinetochore Ska complex. Nature communications, 5, 2964. https://doi.org/10.1038/ncomms3964
Aguilera M., Oliveros M., Martinez-Padron M., Barbas J. A. and Ferrus A. (2000). Ariadne-1: a vital Drosophila gene is required in development and defines a new conserved family of ring-finger proteins. Genetics 155, 1231-1244.
Beacham, D. A., Amatangelo, M. D., & Cukierman, E. (2006). Preparation of extracellular matrices produced by cultured and primary fibroblasts. Current protocols in cell biology, 33(1), 10-9.
Beaulaton, J., & Lockshin, R. A. (1977). Ultrastructural study of the normal degeneration of the intersegmental muscles of Antheraea polyphemus and Manduca sexta (Insecta, Lepidoptera) with particular reference to cellular autophagy. Journal of morphology, 154(1), 39-57.
Bornens, M., & Azimzadeh, J. (2007). Origin and evolution of the centrosome. In Eukaryotic Membranes and Cytoskeleton (pp. 119-129). Springer, New York, NY.
Bratton, S. B., Walker, G., Srinivasula, S. M., Sun, X. M., Butterworth, M., Alnemri, E. S., & Cohen, G. M. (2001). Recruitment, activation and retention of caspases?9 and?3 by Apaf? 1 apoptosome and associated XIAP complexes. The EMBO journal, 20(5), 998-1009.
Burlacu A. (2003). Regulation of apoptosis by Bcl-2 family proteins. Journal of cellular and molecular medicine, 7(3), 249–257. https://doi.org/10.1111/j.1582-4934.2003.tb00225.
Cabal-Hierro L, Lazo PS. Signal transduction by tumor necrosis factor receptors. Cell Signal.2012;24(6):1297–1305.
Cai L, Fritz D, Stefanovic L, Stefanovic B. (2010) Binding of LARP6 to the conserved 5’ stem loop regulates translation of mRNAs encoding type I collagen. J Mol Biol. 395:30926.
Chabin-Brion, K., Marceiller, J., Perez, F., Settegrana, C., Drechou, A., Durand, G., & Pous, C. (2001). The Golgi complex is a microtubule-organizing organelle. Molecular biology of the cell, 12(7), 2047-2060.
Conduit, P. T., Wainman, A., & Raff, J. W. (2015). Centrosome function and assembly in animal cells. Nature reviews Molecular cell biology, 16(10), 611-624.
Davenport, R. W., Dou, P., Rehder, V., & Kater, S. B. (1993). A sensory role for neuronal growth cone filopodia. Nature, 361(6414), 721-724.
Datta, S. R., Katsov, A., Hu, L., Petros, A., Fesik, S. W., Yaffe, M. B., & Greenberg, M. E. (2000). 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Molecular cell, 6(1), 41-51.
Delbridge, A. R., Grabow, S., Strasser, A., & Vaux, D. L. (2016). Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nature reviews Cancer, 16(2), 99.
Dermit, M., Dodel, M., Lee, F., Azman, M. S., Schwenzer, H., Jones, J. L., Blagden, S. P., Ule, J., & Mardakheh, F. K. (2020). Subcellular mRNA Localization Regulates Ribosome Biogenesis in Migrating Cells. Developmental cell, 55(3), 298–313.e10. https://doi.org/10.1016/j.devcel.2020.10.006
Dimitratos, S. D., Stathakis, D. G., Nelson, C. A., Woods, D. F., & Bryant, P. J. (1998). The Location of HumanCASKat Xp11. 4 Identifies This Gene as a Candidate for X-Linked Optic Atrophy. Genomics, 51(2), 308-309.
Dong, G., Chakshusmathi, G., Wolin, S. L., & Reinisch, K. M. (2004). Structure of the La motif: a winged helix domain mediates RNA binding via a conserved aromatic patch. The EMBO journal, 23(5), 1000–1007. https://doi.org/10.1038/sj.emboj.7600115
Dumontet, C., Durán, G. E., Steger, K. A., Murphy, G. L., Sussman, H. H., & Sikic, B. I. (1996).
Differential expression of tubulin isotypes during the cell cycle. Cell motility and the cytoskeleton, 35(1), 49-58.
Eckelman, B. P., Salvesen, G. S., & Scott, F. L. (2006). Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO reports, 7(10), 988-994.
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicologic pathology, 35(4), 495-516.
Eskes, R., Desagher, S., Antonsson, B., & Martinou, J. C. (2000). Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Molecular and cellular biology, 20(3), 929-935.
Gardner, K. L., Sanford, J. L., Mays, T. A., & Rafael?Fortney, J. A. (2006). CASK localizes to nuclei in developing skeletal muscle and motor neuron culture models and is agrin? independent. Journal of cellular physiology, 206(1), 196-202.
Ghosh, A. K. (2002). Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Experimental biology and medicine, 227(5), 301-314.
Goldstein, J. C., Waterhouse, N. J., Juin, P., Evan, G. I., & Green, D. R. (2000). The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nature cell biology, 2(3), 156-162.
Green, D. R., & Llambi, F. (2015). Cell death signaling. Cold Spring Harbor perspectives in biology, 7(12), a006080.
Groban, E. S., Narayanan, A., & Jacobson, M. P. (2006). Conformational changes in protein loops and helices induced by post-translational phosphorylation. PLoS Comput Biol, 2(4), e32.
Guetg, C., & Santoro, R. (2012). Formation of nuclear heterochromatin: the nucleolar point of view. Epigenetics, 7(8), 811-814.
Gunning, P. W., Ghoshdastider, U., Whitaker, S., Popp, D., & Robinson, R. C. (2015). The evolution of compositionally and functionally distinct actin filaments. J Cell Sci, 128(11), 2009-2019.
Hau, H., Ogundele, O., Hibbert, A. H., Monfries, C., Exelby, K., Wood, N. J., Nevarez-Mejia, J., Carbajal, M. A., Fleck, R. A., Dermit, M., Mardakheh, F. K., Williams-Ward, V. C., Pipalia, T. G., Conte, M. R., & Hughes, S. M. (2020). Maternal Larp6 controls oocyte development, chorion formation and elevation. Development (Cambridge, England), 147(4), dev187385. https://doi.org/10.1242/dev.187385
Hsueh, Y. P. (2006). The role of the MAGUK protein CASK in neural development and synaptic function. Current medicinal chemistry, 13(16), 1915-1927.
Hutt, K. J. (2015). The role of BH3-only proteins in apoptosis within the ovary. Reproduction, 149(2), R81-R89.
Imai, R., Nozaki, T., Tani, T., Kaizu, K., Hibino, K., Ide, S., ... & Maeshima, K. (2017). Density imaging of heterochromatin in live cells using orientation-independent-DIC microscopy. Molecular biology of the cell, 28(23), 3349-3359.
Irmler, M., Thome, M., Hahne, M., Schneider, P., Hofmann, K., Steiner, V., ... & Rimoldi, D. (1997). Inhibition of death receptor signals by cellular FLIP. Nature, 388(6638), 190-195.
Jensen, E. (2014). Technical review: colocalization of antibodies using confocal microscopy. The Anatomical Record, 297(2), 183-187.
Jiang, Peng et al. “The Bad guy cooperates with good cop p53: Bad is transcriptionally up- regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis.” Molecular and cellular biology vol. 26,23 (2006): 9071-82. doi:10.1128/MCB.01025-06
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell. 2010;141(1):52-67. https://www.sciencedirect.com
Kahn, O. I., & Baas, P. W. (2016). Microtubules and growth cones: motors drive the turn. Trends in neurosciences, 39(7), 433-440.
Kim, H. E., Du, F., Fang, M., & Wang, X. (2005). Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proceedings of the National Academy of Sciences, 102(49), 17545-17550.
Linhoff, M. W., Garg, S. K., & Mandel, G. (2015). A high-resolution imaging approach to investigate chromatin architecture in complex tissues. Cell, 163(1), 246-255.
Lockshin RA, and Williams CM. (1965) Programmed Cell Death--I. Cytology Of Degeneration In The Intersegmental Muscles Of The Pernyi Silkmoth. J Insect Physiol. 1965 Feb;11:123-33.
Lodish, H., Berk, A., Zipursky, S. L., Matsudaira, P., Baltimore, D., & Darnell, J. (2000). Molecular cell biology 4th edition. National Center for Biotechnology Information, Bookshelf.
Malladi, S., Challa?Malladi, M., Fearnhead, H. O., & Bratton, S. B. (2009). The Apaf?1• procaspase?9 apoptosome complex functions as a proteolytic?based molecular timer. The EMBO journal, 28(13), 1916-1925.
Manojlovic, Z., & Stefanovic, B. (2012). A novel role of RNA helicase A in regulation of translation of type I collagen mRNAs. Rna, 18(2), 321-334.
Manojlovic, Z., Blackmon, J., & Stefanovic, B. (2013). Tacrolimus (FK506) prevents early stages of ethanol induced hepatic fibrosis by targeting LARP6 dependent mechanisms of collagen synthesis. PLoS One, 8(6).
Marin I. and Ferrus A. (2002) Comparative genomics of the RBR family, including the Parkinson's disease-related gene parkin and the genes of the ariadne subfamily. Mol Biol Evol 19, 2039-2050.
Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget. 2014;5(9):2736-2749. https://www.openaire.eu/search/publication?articleId=od 267::af0bdcc61c88b887 1b194060911a1dbf.
Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190, 2003. doi:10.1016/S0092-8674(03)00521-X.
Ming, X. Y., Fu, L., Zhang, L. Y., Qin, Y. R., Cao, T. T., Chan, K. W., ... & Guan, X. Y. (2016).
Integrin α7 is a functional cancer stem cell surface marker in oesophageal squamous cell carcinoma. Nature communications, 7(1), 1-14.
Miura, M., Zhu, H., Rotello, R., Hartwieg, E. A., & Yuan, J. (1993). Induction of apoptosis in fibroblasts by IL-1β-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell, 75(4), 653-660.
Moldoveanu, T., & Czabotar, P. E. (2020). BAX, BAK, and BOK: a coming of age for the BCL-2 family effector proteins. Cold Spring Harbor Perspectives in Biology, 12(4), a036319.
Moynihan T. P., Ardley H. C., Nuber U., Rose S. A., Jones P. F., Markham A. F., Scheffner M. and Robinson P. A. (1999) The ubiquitin-conjugating enzymes UbcH7 and UbcH8 interact with RING finger/IBR motif-containing domains of HHARI and H7-AP1. J Biol Chem 274, 30963-30968.
Muñoz-Pinedo, C., Guío-Carrión, A., Goldstein, J. C., Fitzgerald, P., Newmeyer, D. D., & Green, D. R. (2006). Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proceedings of the National Academy of Sciences, 103(31), 11573-11578.
Nagata S, Tanaka M. Programmed cell death and the immune system. Nature reviews.
Immunology. 2017;17(5):333-340. https://www.ncbi.nlm.nih.gov/pubmed/28163302. doi: 10.1038/nri.2016.153.
Názer, E., Verdún, R. E., & Sánchez, D. O. (2011). Nucleolar localization of RNA binding proteins induced by actinomycin D and heat shock in Trypanosoma cruzi. PLoS One, 6(5), e19920.
Norbury CJ, Hickson ID. Cellular responses to DNA damage. Annu Rev Pharmacol Toxicol.
2001;41:367–401.
Ojeh, N., Pekovic, V., Jahoda, C., & Määttä, A. (2008). The MAGUK-family protein CASK is targeted to nuclei of the basal epidermis and controls keratinocyte proliferation. Journal of cell science, 121(16), 2705-2717.
Oriola, D., Needleman, D. J., & Brugués, J. (2018). The physics of the metaphase spindle. Annual review of biophysics, 47, 655-673.
Pawley, J. (Ed.). (2006). Handbook of biological confocal microscopy (Vol. 236). Springer Science & Business Media.
Pelletier, J., Thomas, G., & Volarevi?, S. (2018). Ribosome biogenesis in cancer: new players and therapeutic avenues. Nature Reviews Cancer, 18(1), 51-63.
Peng, J., Ding, J., Tan, C., Baggenstoss, B., Zhang, Z., Lapolla, S. M., & Lin, J. (2009).
Oligomerization of membrane-bound Bcl-2 is involved in its pore formation induced by tBid. Apoptosis, 14(10), 1145-1153.
Reed JC. Dysregulation of apoptosis in cancer. Journal of Clinical Oncology. 1999;17(9). Rousseau, J., Lamoureux, F., Picarda, G., Gouin, F., Trichet, V., & Rédini, F. (2010). Animal
Models of Malignant Primary Bone Tumors and Novel Therapeutic Approaches. In Bone cancer (pp. 333-346). Academic Press.
Scaffidi, C., Kirchhoff, S., Krammer, P. H., & Peter, M. E. (1999). Apoptosis signaling in lymphocytes. Current opinion in immunology, 11(3), 277-285.
Schober, J. M., Komarova, Y. A., Chaga, O. Y., Akhmanova, A., & Borisy, G. G. (2007).
Microtubule-targeting-dependent reorganization of filopodia. Journal of cell science, 120(7), 1235-1244.
Shao, R., Scully, S. J., Jr, Yan, W., Bentley, B., Mueller, J., Brown, C., Bigelow, C., & Schwartz,
L. M. (2012). The novel lupus antigen related protein acheron enhances the development of human breast cancer. International journal of cancer, 130(3), 544–554. https://doi.org/10.1002/ijc.26015
Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes & cancer.
2011;2(12):1097-1105. https://www.ncbi.nlm.nih.gov/pubmed/22866201. doi: 10.1177/1947601911423031.
Slee EA, Adrain C, Martin SJ. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem. 2001;276:7320–6.
Sonnen, K. F., Schermelleh, L., Leonhardt, H., & Nigg, E. A. (2012). 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biology open, 1(10), 965-976.
Stefanovic, L., Longo, L., Zhang, Y., & Stefanovic, B. (2014). Characterization of binding of LARP6 to the 5’stem-loop of collagen mRNAs: Implications for synthesis of type I collagen. RNA biology, 11(11), 1386-1401.
Strawbridge, R. J., Dupuis, J., Prokopenko, I., Barker, A., Ahlqvist, E., Rybin, D., ... & Nica, A. (2011). Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes, 60(10), 2624-2634.
Sun, R., Chen, W., Zhao, X., Li, T., & Song, Q. (2011). Acheron regulates vascular endothelial proliferation and angiogenesis together with Id1 during wound healing. Cell biochemistry and function, 29(8), 636-640.
Tapscott SJ, Davis RL, Thayer MJ, Cheng PF, Weintraub H, Lassar AB (Oct 1988). "MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts". Science. 242 (4877): 405–511.
Tait, S. W., & Green, D. R. (2010). Mitochondria and cell death: outer membrane permeabilization and beyond. Nature reviews Molecular cell biology, 11(9), 621-632.
Thomson, E., Ferreira-Cerca, S., & Hurt, E. (2013). Eukaryotic ribosome biogenesis at a glance. Tsujimoto, Y. (1998). Role of Bcl?2 family proteins in apoptosis: apoptosomes or mitochondria?.
Genes to cells, 3(11), 697-707.
Verstraelen, P., Detrez, J. R., Verschuuren, M., Kuijlaars, J., Nuydens, R., Timmermans, J. P., & De Vos, W. H. (2017). Dysregulation of microtubule stability impairs morphofunctional connectivity in primary neuronal networks. Frontiers in cellular neuroscience, 11, 173.
Vinogradova, T., Paul, R., Grimaldi, A. D., Loncarek, J., Miller, P. M., Yampolsky, D., ... & Kaverina, I. (2012). Concerted effort of centrosomal and Golgi-derived microtubules is
required for proper Golgi complex assembly but not for maintenance. Molecular biology of the cell, 23(5), 820-833.
Vukmirovic, M., Manojlovic, Z., & Stefanovic, B. (2013). Serine-threonine kinase receptor- associated protein (STRAP) regulates translation of type I collagen mRNAs. Molecular and cellular biology, 33(19), 3893-3906.
Wajant, H. (2002). The Fas signaling pathway: more than a paradigm. Science, 296(5573), 1635- 1636.
Wang, C., Li, J., Ye, S., Zhang, Y., Li, P., Wang, L., & Wang, T. H. (2018). Oestrogen inhibits VEGF expression and angiogenesis in triple-negative breast cancer by activating GPER-
1. Journal of Cancer, 9(20), 3802.
Wang, H., & Stefanovic, B. (2014). Role of LARP6 and Nonmuscle Myosin in Partitioning of Collagen mRNAs to the ER Membrane. PLoS One, 9(10), e108870.
Wang Z. (2003) Acheron, a novel regulator of myoblast differentiation. PhD Thesis, University of Massachusetts at Amherst.
Weigand, J. E., Boeckel, J. N., Gellert, P., & Dimmeler, S. (2012). Hypoxia-induced alternative splicing in endothelial cells. PloS one, 7(8).
Weng, H., Kim, C., Valavanis, C., Wang, Z., & Schwartz, L. (2009). Acheron, an novel LA antigen family member, binds to CASK and forms a complex with Id transcription factors. Cellular and Molecular Biology Letters, 14(2), 273-287.
Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. doi: 10.1016/1074- 7613(95)90057-8.
William D. Foulkes, M.B., B.S., Ian E. Smith, Jorge S. Reis-Filho. Triple-negative breast cancer.
. . doi: 10.1056/NEJMra1001389.
Yaffe, D., & Saxel, O. (1977). Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature, 270(5639), 725–727. https://doi.org/10.1038/270725a0
Yang, X., Luo, C., Cai, J., Pierce, W. M., & Tezel, G. (2008). Phosphorylation-dependent interaction with 14-3-3 in the regulation of bad trafficking in retinal ganglion cells. Investigative ophthalmology & visual science, 49(6), 2483-2494.
Yuan, S., Yu, X., Topf, M., Ludtke, S. J., Wang, X., & Akey, C. W. (2010). Structure of an apoptosome-procaspase-9 CARD complex. Structure, 18(5), 571-583.
Yu, X., Acehan, D., Ménétret, J. F., Booth, C. R., Ludtke, S. J., Riedl, S. J., ... & Akey, C. W. (2005). A structure of the human apoptosome at 12.8 Å resolution provides insights into this cell death platform. Structure, 13(11), 1725-1735.
Zhang, Y., & Stefanovic, B. (2017). mTORC1 phosphorylates LARP6 to stimulate type I collagen expression. Scientific reports, 7(1), 1-15.
Zou, H., Henzel, W. J., Liu, X., Lutschg, A., & Wang, X. (1997). Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c–dependent activation of caspase-3. Cell, 90(3), 405-413.

- MIS604 Requirement Engineering Case Study
- MIS101 Information Systems for Business
- HLTINF001 Comply With Infection Prevention and Control Assignment
- MGT601 Dynamic Leadership Report
- The Australian Solicitors Conduct Rules
- BSBSUS511 Develop Workplace Policy and Procedure Assignment
- Research and Statistical Methods for Business
- BE486 Organisational Behaviour and Human Resource Management Assignment
- ICT80011 Research Method Report
- LAW3700 International Trade Law Assignment
- NUR5011 Contemporary Nursing Assignment
- NURBN1015 Introduction to Evidence Based Practice and Research Assignment
- PROJ6004 Data Reporting Assignment
- FPH201 First Peoples Culture History and Healthcare Report 1
- PROG2008 Computational Thinking Assignment
- BSBWOR502B Ensure Team Effectiveness Assignment
- AFE4001 Business Economics Assignment
- MIS605 Systems Analysis and Design Case Study 2
- NURBN2024 Mental Health Recovery Plan Case Study
- PBHL20008 Engaging with Cross Cultural Communities Assignment


.png)
~5.png)
.png)
~1.png)






















































.png)






